拉曼光谱内标法定量分析Purex有机体系中的U(Ⅵ)
2015-12-25李定明常志远康海英
白 雪,李定明,常志远,康海英
中国原子能科学研究院 放射化学研究所,北京 102413
BAI Xue,LI Ding-ming,CHANG Zhi-yuan,KANG Hai-ying
China Institute of Atomic Energy,P.O.Box 275(88),Beijing 102413,China
在Purex后处理工艺流程中,有机相体系中U(Ⅵ)的分析点多,分析频率高,其浓度的准确测定与否影响整个工艺的稳定可靠运行,因此建立快速、准确的分析方法非常重要。目前有机相中铀定量分析方法有:滴定法[1-2]、γ吸收法[3]、拉曼光谱法[4-5]等。
滴定法作为一种经典方法,在铀的定量分析领域应用广泛、准确度较高,但是方法繁琐、操作时间长且产生废液难以回收利用,对操作人员的要求较高,难以实现自动化分析。
γ吸收法是目前后处理厂普遍采用的方法,具有很多优点,但也有一定局限性,铀浓度的检测受部分杂质的影响较大,尤其当铀的浓度与杂质浓度比值较低时,分析准确度不高,并且透射源的γ射线的统计涨落也会影响方法的准确度。
拉曼光谱法作为一种无损检测技术,被广泛应用于多种有机物和无机物的定性和定量分析。铀酰离子属于Y—X—Y型线型离子,各种振动模型中只有U O键的ν1对称伸缩振动是拉曼活性的。早在20世纪50年代,Sutton[6]就通过拉曼光谱法研究了的结构。此后关于的拉曼光谱研究大多数集中于定性分析[7-8]。美国西北太平洋国家实验室的Bryan等[9-10]将拉曼光谱法应用于水相中的定量分析,并对Purex流程水相料液的U(Ⅵ)进行拉曼光谱在线监测。在硝酸铀酰有机相体系中,的ν1对称伸缩振动峰位于860cm-1,可利用860cm-1处的拉曼信号强度与其浓度的线性关系,来测定有机相中的U(Ⅵ)浓度[11]。拉曼光谱法测定铀浓度简便快捷、绿色无损、不产生废液,且不受体系中共存的硝酸及U(Ⅳ)等物质的影响。
在一定条件下,拉曼信号的强度与待测物的浓度成正比,但在实际检测应用中,由于受到检测器稳定性、暗电流噪声、样品放置位置等因素的影响,标准曲线可能偏离原有的线性,尤其在长时间检测时偏离更为明显。另外,当改变仪器参数(如积分时间、激光功率等)或检测样品所用的容器时,标准曲线往往不能通用,需要重新绘制标准曲线,这需要耗费更多的时间。通常选择加入内标物来消除标准曲线的偏离,对非水溶液,常选择四氯化碳为内标,但是额外加入内标物对放射性体系不利,除了增加操作步骤外,也会产生放射性废液难以分离回收。
针对以上问题,本工作提出使用有机体系中的溶剂作为内标物的方法,无需另外加入内标物,以简化实验步骤,且检测方法不产生废液、绿色无损。
1 实验部分
1.1 仪器与试剂
iRaman便携式拉曼光谱仪,B&WTEK公司,激光波长785nm,激光功率300mW,探头工作距离为5mm,光纤长度为10m。
U3O8(GBW04205),北京化工冶金研究院;硝酸、磷酸三丁酯(TBP),分析纯,国药集团化学试剂有限公司;240#加氢煤油,锦州化工厂。
30%TBP/煤油,按体积配制,使用前经5%Na2CO3溶液处理。
U3O8经浓HNO3加热溶解配制硝酸铀酰水相溶液,在高浓度硝酸条件下由30%TBP/煤油萃取铀水相母液得到有机相母液,采用TiCl3还原-K2Cr2O7氧化滴定法标定U(Ⅵ)浓度,近红外光谱法标定HNO3浓度。不同浓度硝酸铀酰有机相溶液由其母液稀释配制。
1.2 实验方法
1.2.1 溶剂的辐照实验 30%TBP/煤油与等体积3.0mol/L硝酸混合震荡后,静置分离,取有机相置于玻璃辐照管中,在不同的剂量(104、105、106Gy)下进行辐照。照射源为2.89×1015Bq的60Co放射源,用重铬酸银剂量计测得辐照点的剂量率为3 500Gy/h。
1.2.2 标准曲线的绘制 将不同浓度硝酸铀酰有机溶液置于比色池中,采集其拉曼光谱,积分时间7s,平均次数2次,激光功率100%。将860cm-1处U(Ⅵ)的拉曼信号强度对其浓度作图,得到未使用内标法时的标准曲线。扣除拉曼光谱的荧光背景后,使用833、891、1 065、1 302cm-1的四个峰作为内标峰,拉曼光谱除以内标峰的高度,得到以相对强度为纵坐标的拉曼光谱,以860cm-1处U(Ⅵ)的拉曼信号相对强度对其浓度作图,得到使用内标法时的标准曲线。以上步骤在75d后重复进行一次,以确定标准曲线随时间的变化。
2 结果与讨论
2.1 内标物的拉曼光谱稳定性
在Purex后处理工艺流程中,有机体系的溶剂为30%TBP/煤油,其浓度相对固定且具有较好的热稳定性、化学稳定性及辐照稳定性。但是溶剂仍能发生酸催化水解或射线辐照分解,TBP分解生成磷酸二丁酯(DBP)、磷酸一丁酯(MBP)、H3PO4等,煤油分解生成烷基酸、硝基烷、硝酸酯等,会影响有机溶剂的组成和配比[12]。
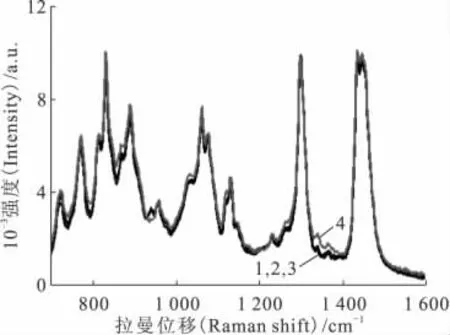
图1 辐照前后30%TBP/煤油的拉曼光谱Fig.1 Raman spectra of 30%TBP/kerosene before and after irradiation
30%TBP/煤油酸化后其主要拉曼谱峰高度及位置未发生改变,而酸化后的有机溶剂更易辐解。为了研究部分降解后有机溶剂拉曼光谱的变化,采集辐照不同剂量后酸化溶剂的拉曼光谱,并与未经辐照的拉曼光谱进行对比,结果示于图1。辐照剂量为104、105Gy时,溶剂拉曼光谱与未经辐照的基本重合;而辐照剂量为106Gy时,溶剂的颜色由无色变为淡黄色,为溶剂与多种降解产物的混合物,有机溶剂的组成及配比均发生了改变,但其拉曼光谱除了基线略微变化外,主要谱峰的位置及高度均未发生明显改变。说明拉曼光谱对溶剂的降解不敏感,溶剂组成及浓度的小范围涨落不影响其拉曼光谱的形状及位置,故可以利用其拉曼特征谱峰作为内标,对有机溶剂中的U(Ⅵ)浓度进行定量分析。
2.2 内标峰的选择
30%TBP/煤油有多个拉曼特征峰,选择内标峰时需满足以下要求:内标峰与U(Ⅵ)的拉曼特征谱峰(860cm-1)不能重合或相隔太远;与U(Ⅵ)的特征拉曼谱峰高度不要相差太多;峰的形状要尖锐等。因此初步选择833、891、1 065、1 302cm-1的四个峰作为内标峰。图2为硝酸铀酰有机溶液的拉曼光谱,图中标示出U(Ⅵ)的特征峰及四个内标峰。
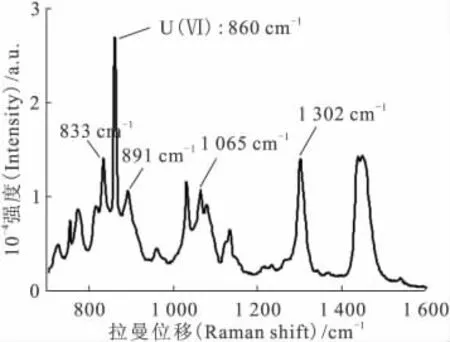
图2 硝酸铀酰有机溶液的拉曼光谱Fig.2 Raman spectrum of uranyl nitrate in 30%TBP/kerosene
分别以四个溶剂峰做内标峰,绘制不同浓度的U(Ⅵ)有机溶液的标准曲线。首先使用采谱软件自带的背景扣除功能,扣除拉曼光谱的荧光背景,再以特征峰(860cm-1)强度与内标峰强度的比值作为相对强度,将相对强度对U(Ⅵ)浓度作图,得到标准曲线。四组内标峰对应的标准曲线示于图3,拉曼信号相对强度与U(Ⅵ)的浓度均呈现良好的线性关系,r2均为0.999,说明有机溶剂的四个拉曼特征峰均有望作为体系中U(Ⅵ)检测的内标峰。
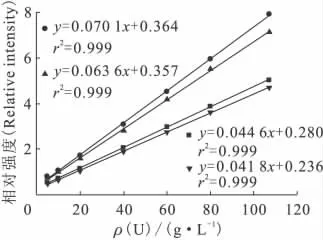
图3 使用不同内标峰的U(Ⅵ)检测标准曲线Fig.3 Calibration curves for uranyl detection using different internal standard peaks
分别以溶剂的四个拉曼特征峰为内标峰,比较标准曲线随时间的稳定性。相隔75d时,分别采集不同浓度硝酸铀酰有机溶液的拉曼光谱,并使用上述四个内标峰绘制标准曲线。标准曲线随时间的变化示于图4。F检验说明在显著性水平α=0.05时,图4(a)中两条直线的分析精度存在显著性差异。t检验显示在显著性水平α=0.05时,图4(a)、(b)、(d)中两条直线的斜率均存在显著性差异。即相隔75d之后,以833、891、1 302cm-1处的拉曼峰为内标峰的标准曲线发生了偏移;而以1 065cm-1拉曼峰为内标峰的标准曲线基本未发生改变。说明在实际应用中,以1 065cm-1处有机溶剂的拉曼特征峰为内标峰时,标准曲线呈现良好的时间稳定性,故内标峰选择1 065cm-1处有机溶剂的拉曼峰。
煤油与TBP分别在1 065cm-1与1 062cm-1处有拉曼谱峰,两者峰高接近,拉曼光谱仪无法分辨两个峰,故内标峰为两者的叠加。图5为TBP体积分数在10%~50%之间变动时,TBP/煤油的拉曼光谱变化。833、891、1 302cm-1处谱峰高度随着TBP体积分数的改变而发生明显的变化,五种配比在上述三个位置的谱峰高度的sr(n=5)分别为28.7%、9.3%、6.0%,而1 065cm-1处谱峰高度的sr(n=5)为2.8%。说明1 065cm-1处峰高度随TBP/煤油配比的改变不明显,故使用该峰为内标峰时,无需预先确定溶剂的具体配比。
综上所述,选择有机溶剂位于1 065cm-1处的拉曼峰为内标峰,标准曲线呈现良好的时间稳定性,故在长时间检测时无需重新绘制标准曲线,简化了操作步骤;该内标峰对TBP/煤油体积比的变化不敏感,故在实际检测中,无需预先测得两者的准确体积比。
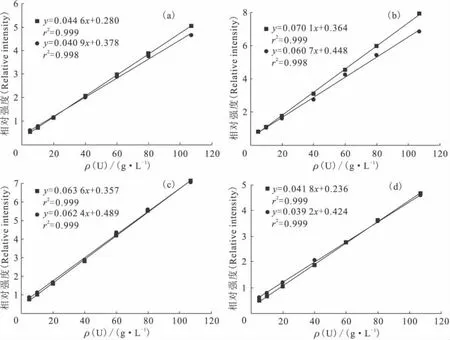
图4 使用不同内标峰时标准曲线的时间稳定性Fig.4 Stability of the calibration curves with different internal standard peaks
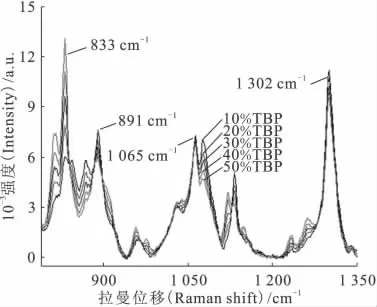
图5 拉曼光谱随TBP/煤油配比的改变Fig.5 Change of Raman spectra with the ratio of TBP and kerosene
2.3 不同因素对内标法定量分析U(Ⅵ)的影响
(1)不同日期的光谱稳定性
在拉曼光谱检测过程中,由于激光光强的变化、检测器稳定性、暗电流噪声、样品放置位置等因素的影响,在不同日期绘制的标准曲线存在一定的差别。图6为相隔75d时绘制的两条标准曲线,纵坐标为拉曼光谱强度,两条标准曲线存在明显的差别;而由图4(c)可知内标峰位置为1 065cm-1时,内标法可以有效消除标准曲线随着时间的偏离。
配制U(Ⅵ)质量浓度为101.0g/L的有机溶液为待测液,在某天采集其拉曼光谱,并将该天设置为时间起点,在1、2、3、7、75d后分别采集待测液的拉曼光谱,采集过程保证仪器参数一致,结果示于图7(a)。不同日期采集到的待测液拉曼光谱峰位置固定,但高度存在明显的差别,860cm-1处谱峰高度的相对标准偏差sr(n=6)为14.4%。而使用内标法后的拉曼光谱相对强度示于图7(b),不同日期采集到的待测液拉曼信号的sr(n=6)降至1.9%,说明内标法能有效消除拉曼光谱随着时间的变化。
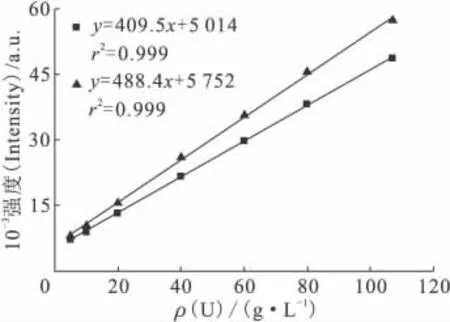
图6 未使用内标法时标准曲线随时间的偏离Fig.6 Change of calibration curves as time without internal standard method
在时间起点当天,采集一系列不同浓度硝酸铀酰有机溶液的拉曼光谱,将U(Ⅵ)拉曼信号强度与相对强度分别对U(Ⅵ)浓度作图,得到两条标准曲线,分别为未使用内标法和使用内标法的情况。由标准曲线与待测样品拉曼光谱对其进行定量分析。
未使用内标法时,由标准曲线及时间起点当天待测液的拉曼光谱计算其质量浓度为99.1g/L,与实际值101.0g/L的相对误差为-1.9%。而其后1、2、3、7、75d测得U(Ⅵ)的质量浓度分别为96.7、95.3、92.0、80.3、128.7g/L,与实际值101.0g/L的相对误差分别为-4.3%、-5.6%、-8.9%、-20.5%、27.4%,结果列于表1。说明在长期检测过程中标准曲线会发生偏离,故为了保证分析结果的准确可靠,每次检测需要重新绘制标准曲线,步骤较为繁琐。
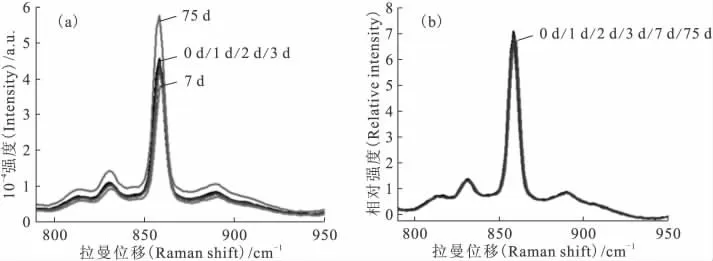
图7 内标法对拉曼光谱随时间变化的消除Fig.7 Effect of internal standard method on the change of Raman spectra as time
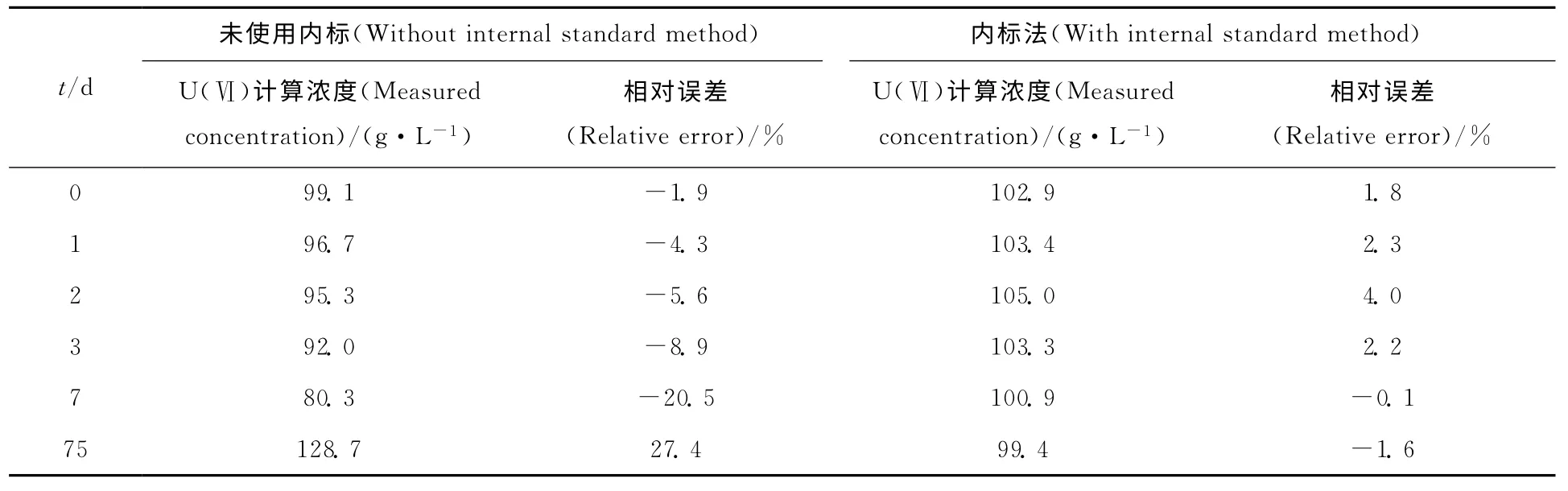
表1 内标法对工作曲线随时间偏移的消除Table 1 Effect of internal standard method on the change of calibration curves as time
使用内标法后,根据标准曲线及待测液的拉曼光谱,测得时间起点与其后1、2、3、7、75d的U(Ⅵ)质 量 浓 度 分 别 为102.9、103.4、105.0、103.3、100.9、99.4g/L,与实际值的相对误差分别为1.9%、2.4%、4.0%、2.3%、-0.1%、-1.6%(表1)。说明本方法可以有效消除标准曲线随着时间的偏离,在每次检测时无需重新绘制标准曲线,大大简化了实验步骤。
(2)容器的影响
放射性样品的分析过程中,样品的分装、转移、制备较为复杂繁琐,常规分析会产生放射性废液和固体废物,如样品不需转移分装即可直接进行浓度测定,得到准确的分析结果,则可简化样品分析步骤,减少废物产生量,实现样品的无损简便分析。
为了研究容器对U(Ⅵ)定量检测的影响,选择5种不同的透明容器,分别为跑兔瓶、比色池、容量瓶、蓝盖瓶和铝盖瓶,容器的材质与形状、容器壁的厚度各有不同。比色池材质为石英,其余4种材质为玻璃。比色池为立方体,探头接触面为平面;容量瓶的探头接触面为球面;而其余3种容器为圆柱体,探头接触面为弧面。5种容器的厚度各有不同,其中蓝盖瓶的厚度远远大于其他容器,5种容器的对比示于图8。
将U(Ⅵ)质量浓度为101.0g/L的有机待测液分别置于5种不同的容器中,光纤探头发出的激光透过容器壁聚焦在待测样中,采集5份待测样的拉曼光谱,并由标准曲线计算其浓度。由于考虑到容器形状与壁厚的不均匀性,跑兔瓶、容量瓶、蓝盖瓶及铝盖瓶均更换3个不同部位进行光谱采集。

图8 5种分析容器Fig.8 Five containers for detection of uranyl
未使用内标法时,利用采集到待测液的拉曼信号强度与标准曲线,可以得到各容器中的样品浓度,结果列于表2。由于绘制标准曲线时使用比色池,故计算得到比色池中待测物浓度为99.1g/L,与实际值101.0g/L的相对误差为-1.9%。分别在跑兔瓶、容量瓶、蓝盖瓶、铝盖瓶三个不同部位采集拉曼光谱,得到待测物浓度平均值与实际值的相对误差为-33.8%、-13.2%、-79.1%、-8.6%。
使用1 065cm-1处的溶剂峰作为内标峰,由不同容器中待测液的拉曼光谱与标准曲线可得到U(Ⅵ)浓度,结果列于表3。由表3结果看出,跑兔瓶、容量瓶、蓝盖瓶、铝盖瓶中U(Ⅵ)的平均质量浓度分别为100.4、101.2、97.2、102.3g/L,与实际值101.0g/L的相对误差分别为-0.6%、0.2%、-3.7%、1.3%,远远优于未使用内标法的情况。
比较使用内标法前后5种容器中待测液的拉曼光谱,结果示于图9。由图9(a)可以看出,未使用内标法时,5组拉曼光谱的强度差别较大,尤其是蓝盖瓶中待测液的拉曼光谱,远远低于比色池中待测液的拉曼光谱。而由图9(b)可以看出,经过内标法进行处理后的拉曼光谱,5组光谱基本重合,说明内标法可以有效消除容器对拉曼光谱的影响,容器不影响浓度定量分析,故检测过程无需进行样品的分装转移,可以简化分析操作、减少放射性废物。
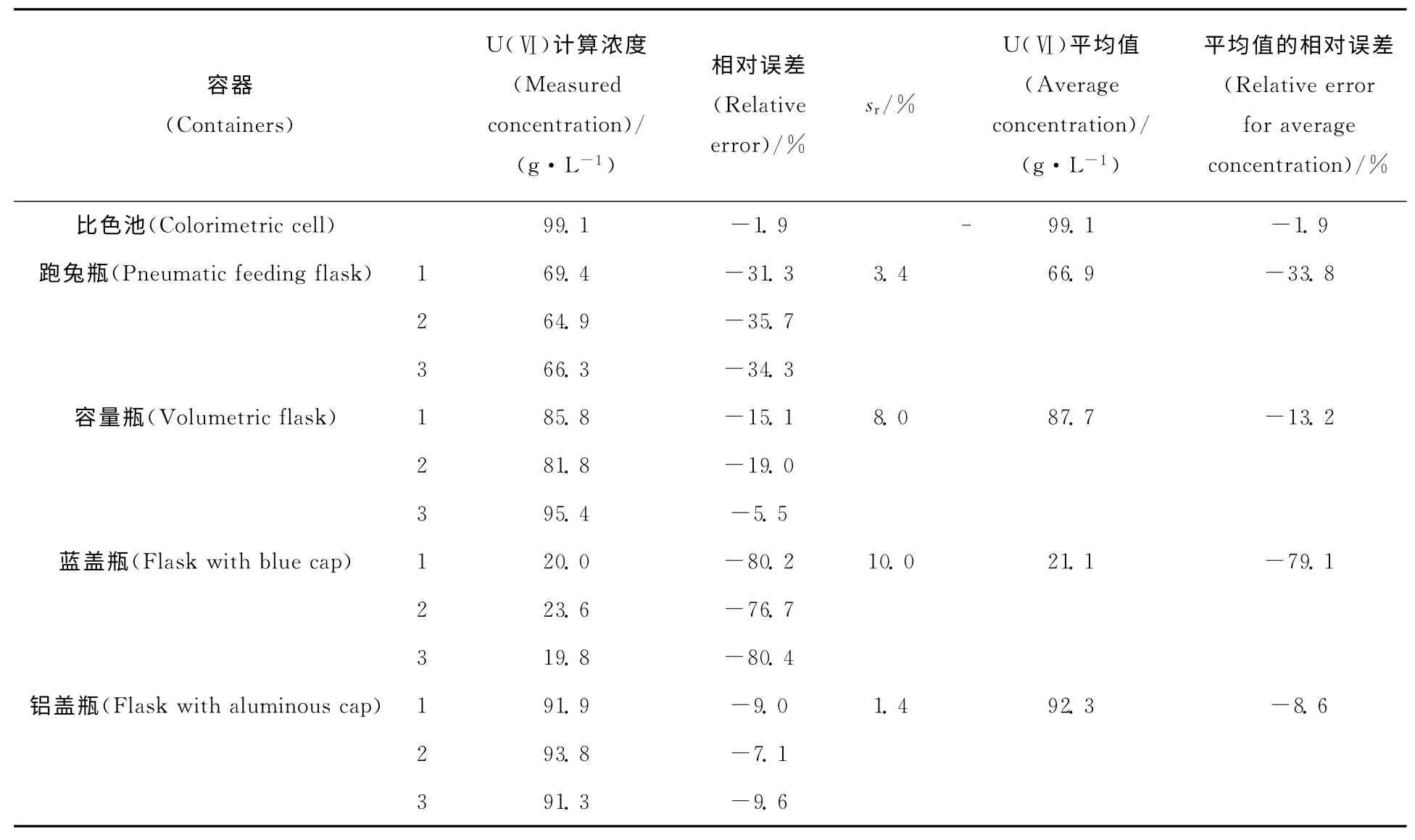
表2 未使用内标法时不同容器中待测液的定量分析结果Table 2 Detection results in different containers without internal standard method
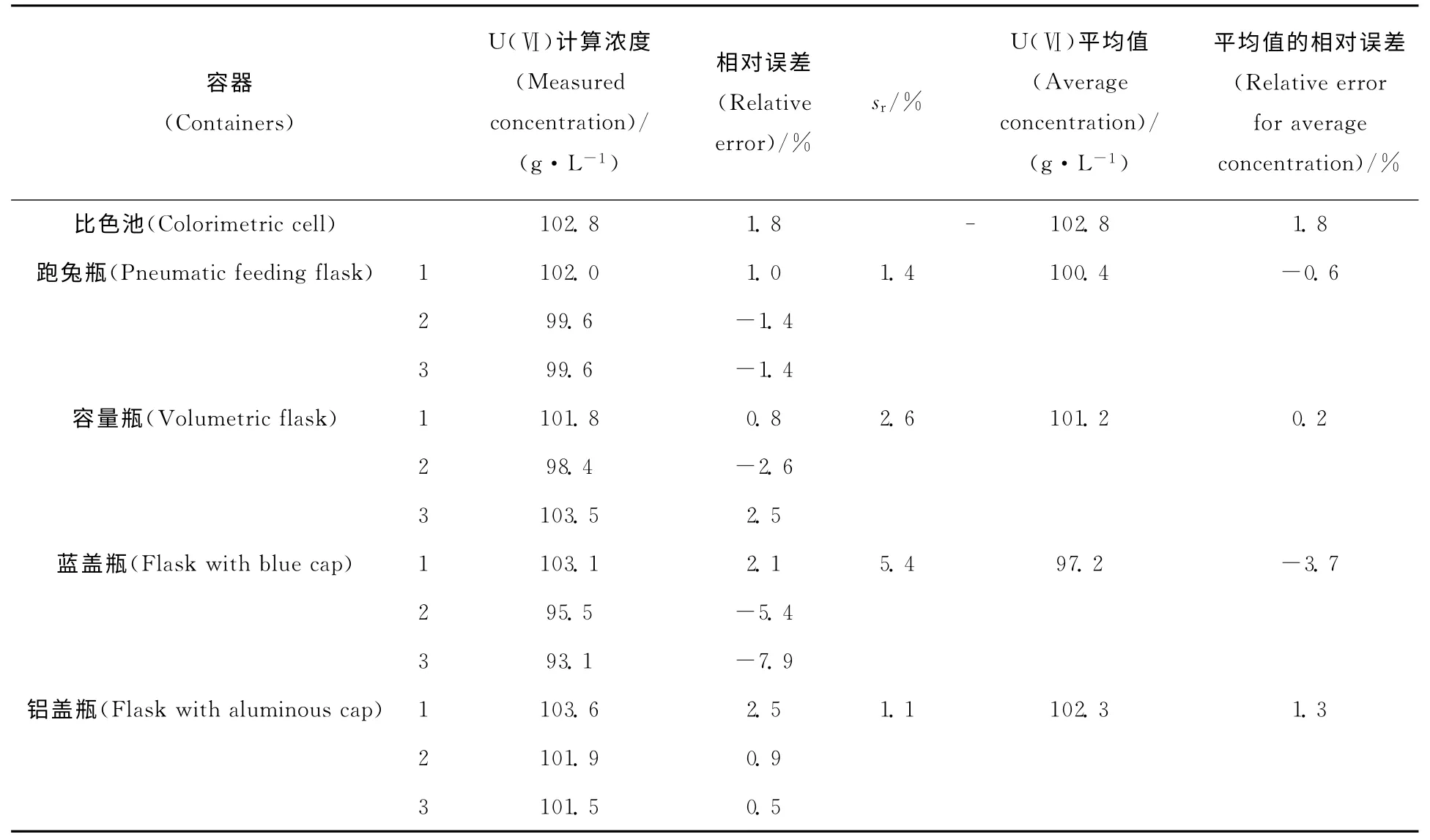
表3 内标法对不同容器中待测液的定量分析结果Table 3 Detection results in different containers with internal standard method
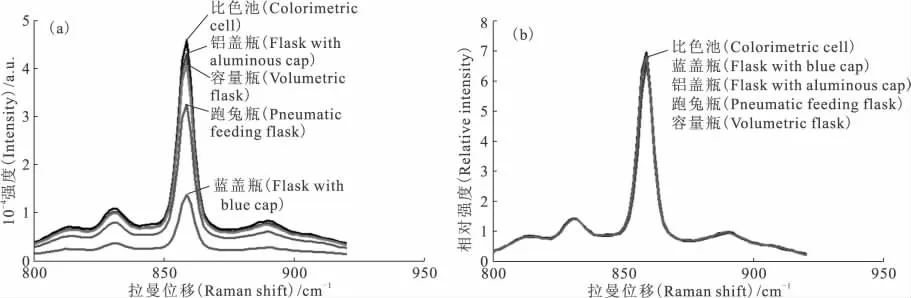
图9 内标法对拉曼光谱随容器影响的消除Fig.9 Effect of internal standard method on the Raman spectra of uranyl in different containers
(3)积分时间的影响
增加拉曼光谱仪的积分时间,可以增加待测物的拉曼信号强度,在分析检测低浓度铀溶液时更为有利。图10为同一份硝酸铀酰有机溶液在不同积分时间下的拉曼光谱对比,积分时间的增加使得UO2+2位于860cm-1处的拉曼信号明显增加,更有利于对其浓度进行准确的分析定量。而分析高浓度铀溶液时,由于UO2+2的拉曼信号较高,需要选择较短的积分时间以确保拉曼信号不超出检测量程。
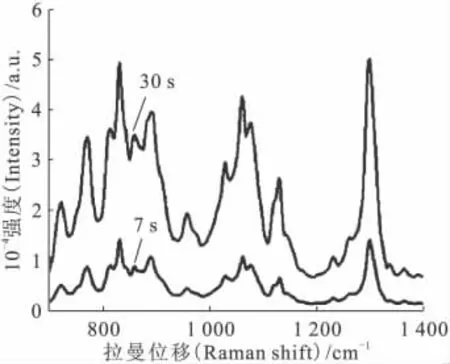
图10 积分时间对U(Ⅵ)拉曼光谱的影响Fig.10 Effect of integral time on the Raman spectra of uranyl
以U(Ⅵ)质量浓度为101.0g/L的硝酸铀酰有机溶液为待测样,在不同的积分时间下采集其拉曼光谱,结果示于图11(a)。由图11(a)可以看出,随着积分时间的增加,860cm-1处U(Ⅵ)的拉曼信号增加,同时拉曼光谱的背景也会增高。使用内标法对光谱进行数据处理,结果示于图11(b),由图11(b)可知,4张光谱基本重合,说明内标法能有效消除积分时间对拉曼光谱的影响。
内标法对积分时间影响的计算结果列于表4。由表4可以看出,使用内标法将拉曼光谱数据处理后,按照标准曲线计算U(Ⅵ)的浓度,分别为94.5、100.7、105.6、102.8g/L,与实际浓度的相对误差分别为-6.4%、-0.3%、4.5%、1.8%。说明按照上述方法可以有效消除积分时间的差异对实验结果造成的影响,在分析检测时可根据U(Ⅵ)浓度的不同选择不同的积分时间:U(Ⅵ)浓度较低时可以选择较长的积分时间,从而使信号更加准确;U(Ⅵ)浓度较高时可以选择较短的积分时间,从而使拉曼信号不超出最大量程。
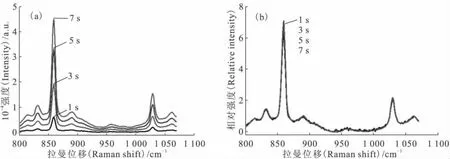
图11 内标法对不同积分时间拉曼光谱的影响Fig.11 Effect of internal standard method on the Raman spectra of uranyl with different integral time
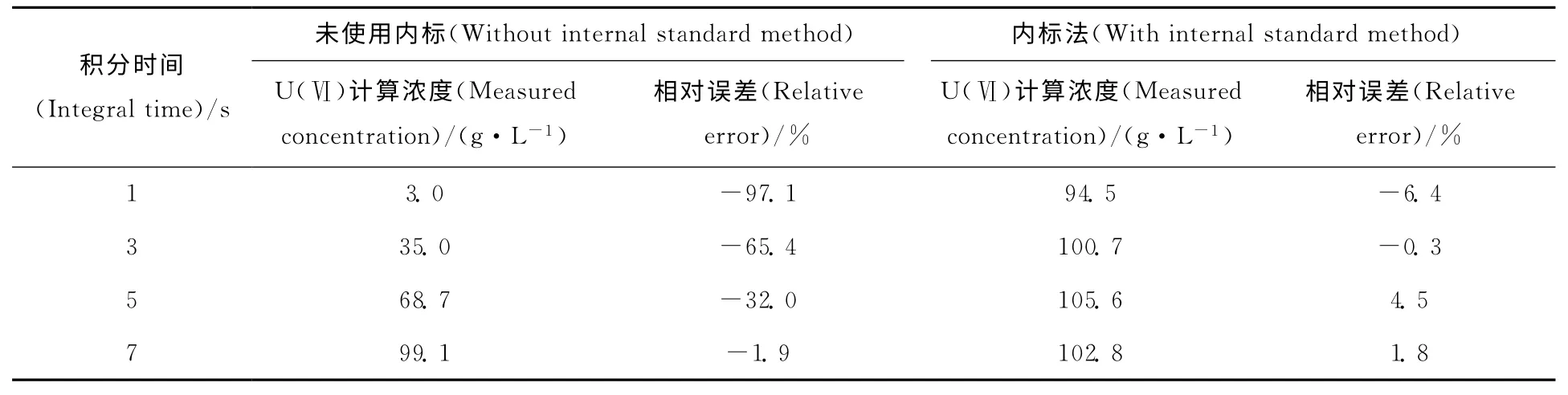
表4 内标法对不同积分时间时U(Ⅵ)的计算结果Table 4 Detection results of U(Ⅵ)with internal standard method with different integral time
(4)激光功率的影响
在改变待测物的拉曼信号强度方面,改变拉曼光谱仪的激光功率与积分时间作用基本相同,分析U(Ⅵ)样品时可以按照U(Ⅵ)浓度的不同选择不同的激光功率,故需考察内标法对激光功率影响的消除。
改变拉曼光谱仪的激光功率,分别采集待测样的拉曼光谱,结果示于图12(a),随着激光功率的增强,位于860cm-1的拉曼特征峰增强。而使用内标法处理后的拉曼光谱示于图12(b),4组拉曼光谱基本重合。
内标法对激光功率影响的计算结果列于表5。由表5可以看出,使用内标法之后,根据标准曲线计算得到U(Ⅵ)的质量浓度分别为103.7、101.7、100.6、102.8g/L,与实际值101.0g/L的相对误差分别为2.7%、0.7%、-0.4%、1.8%。说明内标法可以有效消除激光功率的差异对实验结果造成的影响。在检测U(Ⅵ)有机样品时,可以按照不同的U(Ⅵ)浓度选择不同的激光功率,并使用内标法消除激光功率差异的影响,得到相对准确的分析结果。
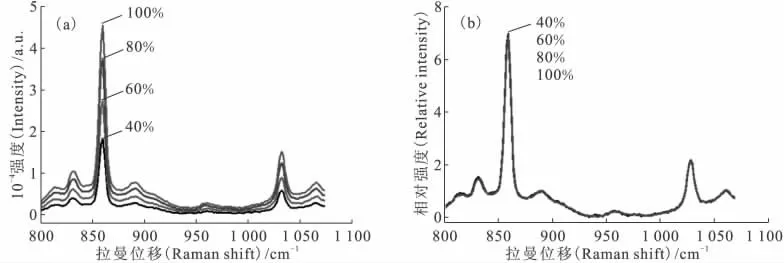
图12 内标法对不同激光功率拉曼光谱的影响Fig.12 Effect of internal standard method on the Raman spectra of uranyl with different laser power
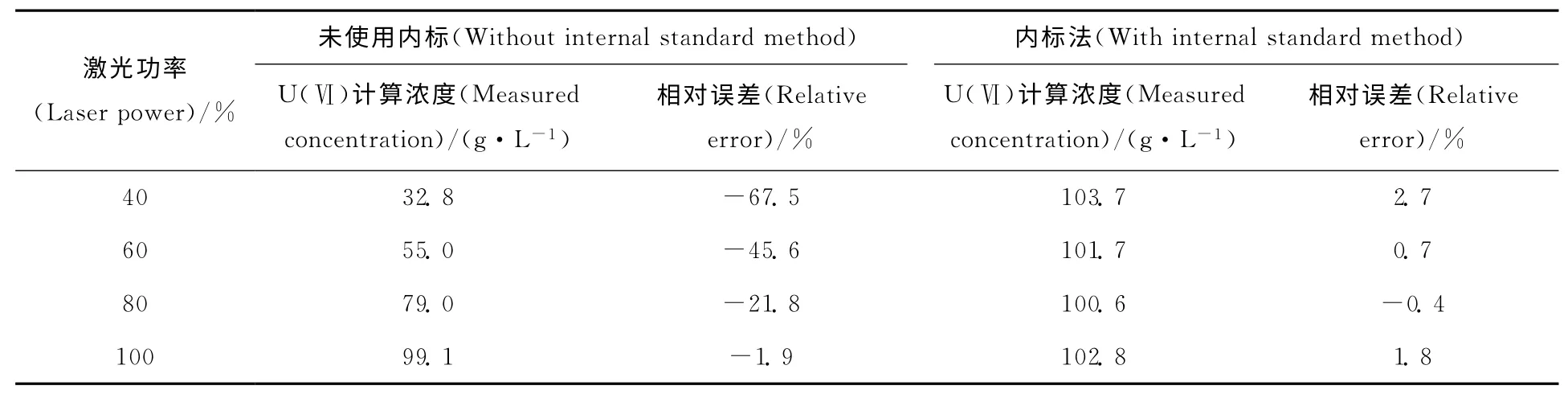
表5 内标法对不同激光功率时U(Ⅵ)的计算结果Table 5 Detection results of U(Ⅵ)with internal standard method with different laser power
3 结 论
在TBP/煤油有机体系中定量检测U(Ⅵ)时,扣除待测物拉曼信号的荧光背景后,使用1 065cm-1处溶剂的拉曼谱峰作为内标峰,U(Ⅵ)的拉曼信号与之相比得到相对强度。拉曼信号相对强度-U(Ⅵ)浓度呈现良好的线性关系,表明该方法可用于Purex工艺有机体系中U(Ⅵ)的定量检测。该检测方法使得标准曲线具有良好的时间稳定性,在75d内标准曲线基本不发生改变,从而检测时无需重新绘制标准曲线,简化了实验步骤。1 065cm-1的内标峰高对TBP/煤油的配比变化不敏感,故实际检测时无需预先确定两者的准确配比。使用该内标峰时,在不同容器中检测到的结果基本相同,故检测过程中无需进行样品的转移及分装,有利于进行无损分析,减少放射性废物产生量。改变拉曼光谱仪的积分时间和激光功率基本不影响U(Ⅵ)的定量检测,从而可选择合适的参数以适应不同浓度U(Ⅵ)溶液分析的需要。内标法增加了标准曲线的通用性,但是在改变某些仪器参数时,测量误差略大,故在实际检测过程中应尽量选择统一的仪器参数。
[1]Wahlberg J S,Skinner D L,Jr Rader L F.Volumetric determination of uranium[J].Anal Chem,1957,29(6):954-957.
[2]Chadwick P H,McGowan I R.Determination of plutonium and uranium in mixed oxide fuels by sequential redox titration[J].Talanta,1972,19:1335-1348.
[3]董焱武,由文职,周其荣,等.流线分析[G]∥1977年流线分析会议资料选编.北京:原子能出版社,1978:147.
[4]Bryan S A,Levitskaia T G,Johnsen A M,et al.Spectroscopic monitoring of spent nuclear fuel reprocessing streams:an evaluation of spent fuel solutions via Raman,visible,and near-infrared spectroscopy[J].Radiochim Acta,2011,99:563-571.
[5]Burck J.Spectrophotometric determination of uranium and nitric-acid by applying partial least-squares regression to uranium(Ⅵ)absorption-spectra[J].Anal Chim Acta,1991,254(1/2):159.
[6]Sutton J.Configuration of the uranyl ion[J].Nature,1952,169:235-236.
[7]Guillaume B,Begun G M,Hahn R L.Raman spectrometric studies of cation-cation complexes of pentavalent actinides in aqueous perchlorate solutions[J].Inorg Chem,1982,21(3):1159-1166.
[8]Toshiyuki F,Kenso F,Hajimu Y,et al.Raman spectroscopic determination of formation constant of uranyl hydrolysis species(UO2)2[J].J Alloy Comp,2001,323-324:859-863.
[9]Bryan S A,Levitskaia T G,Johnsen A M,et al.Spectroscopic monitoring of spent nuclear fuel reprocessing streams:an evaluation of spent fuel solutions via Raman,visible,and near-infrared spectroscopy[J].Radiochim Acta,2011,99:563-571.
[10]Schwantes J M,Bryan S A,Orton C R,et al.Advanced process monitoring safeguards technologies at Pacific Northwest National Laboratory[J].Procedia Chem,2012,7:716-724.
[11]白雪,李定明,常志远,等.铀(Ⅵ)的拉曼光谱定量分析[J].核化学与放射化学,2014,36(S0):27-34.
[12]李辉波,苏哲,丛海峰,等.30%TBP-煤油-硝酸体系的α和γ辐照行为[J].核化学与放射化学,2012,34(5):281-285.
