乏燃料后处理湿法工艺技术基础研究发展现状
2015-12-25张生栋严叔衡
张生栋,严叔衡
1.中国原子能科学研究院 放射化学研究所,北京 102413;2.中国核工业集团公司 科技委,北京 100822
利用商用裂变反应堆发电的50多年以来,核能已发展成为一种可替代化石燃料的基荷性清洁能源。截止2012年12月31日全世界在运行的核电反应堆435台,总装机容量374.108GWe,已有1.4万个堆年的运行经验,提供全球14%的电力需求。尽管日本福岛核事故无疑给全球核电复苏态势带来巨大冲击,一定时期内核电复苏步伐将会有所放缓,但不可能出现上两次核事故后长达20~30a的萧条期。目前主要核电国家均宣称要继续保有核电,美、法、英等老牌核电国家表示坚持核能发展立场。从长远看,经济发展对能源的需求,以及应对全球气候变暖必须减排CO2将是核电发展的两大根本动力,必然会持续推动核电较快地发展。
为了保持核能可持续发展,必须相应发展乏燃料后处理技术,以实施快堆闭合核燃料循环。这不仅是因为快堆比热堆循环可提高铀资源利用率30~60倍[1],使已知的铀资源能够使用几千年[2],而且可实现放射性废物最小化,降低放射性废物的放射毒性和释热量,使放射性废物地质处置库安全监管时间缩短至1 000年[3]7-10。因此,乏燃料后处理是闭合核燃料循环的核心环节,它是核能可持续发展的关键。
乏燃料湿法后处理工艺(PUREX流程)是核武器国家为了生产武器级钚(240Pu含量≤7%的Pu)而发展起来的。随着20世纪60年代核电的发展,为了充分利用铀资源,将军用后处理技术改进和发展用于处理动力堆乏燃料,分离和回收U和Pu用于再循环,产生的高放废液经玻璃固化变成固化体,中间贮存适当时间再进行深地质处置。经过50多年的发展,PUREX流程已是动力堆乏燃料后处理工业规模分离U和Pu过程的基础,目前法、英、俄、日、印等国家已建成了商业乏燃料后处理能力5 575tHM/a[4]的后处理厂。但这种只分离乏燃料中U和Pu的所谓标准的PUREX流程,产生的玻璃固化高放废物中含有全部次锕系元素(MA,如Np、Am、Cm等)和裂变产物(FP),要衰变到天然铀矿的放射性水平将需要1万年以上[5-6]。为此,许多国家正在研发先进的湿法后处理工艺,首先优化PUREX流程的参数,从热堆乏燃料中分离出U、Pu、Np和Tc,然后再从萃余液中分离出长期放射性毒性的MA和短期释热的FP(90Sr和137Cs),使产生的高放玻璃固化体所需的深地质处置库容减少,安全监管时间缩短至1 000年。
从目前国际上后处理技术发展来看,湿法后处理工艺仍以PUREX流程为基础,从乏燃料元件首端处理工艺、萃取工艺的简化和无盐调价,同时分离U、Pu、Np和Tc以及高放废液中镧锕分离等方面开展相关研究,以实现废物的最小化。
1 乏燃料元件高温煅烧氧化的首段工艺技术[6-7]研究现状
在乏燃料剪切后,切断的燃料块在含O2的气氛中加热(>480℃),使陶瓷UO2与O2反应生成细粉末状U3O8,随之体积膨胀约30%。此法的优点如下:
1)简化了燃料与包壳的分离程序
氧化挥发法使陶瓷UO2芯块变成细粉末U3O8或UO3,通过旋转式或振动式氧化反应器的搅拌,可回收大于99%的燃料,少量(<1%)燃料细粉粘附在包壳表面,用HNO3洗涤可以回收。
2)加速了U的溶解速度,减少了HNO3的消耗量,提高U的溶解率
燃料粒径减小可大大地加速HNO3的溶解速度。U的氧化态提高,溶解时释放的NOx量和消耗的HNO3量减少(如表1所示)。产生的UO3粉末很快溶解于浓度大于0.3mol/L HNO3中只产生微量的NOx。
3)加热氧化可从燃料芯块中除去挥发性物质(如Xe、Kr、3H、14C等)和半挥发性物质(如I、Mo、Tc等)。
乏燃料中的氚约有40%以上是在Zr合金包壳中(ZrTx),按理氢化物应该容易氧化释放出氚,但实验结果表明,在480℃下氧化6h,保留在Zr合金包壳中的氚释放较少。但UO2芯块中的氚,在480℃下氧化4h释放率大于99.9%,以HTO形式进入气相中,以免后续湿法分离系统中氚化水的积累,排放到环境中。
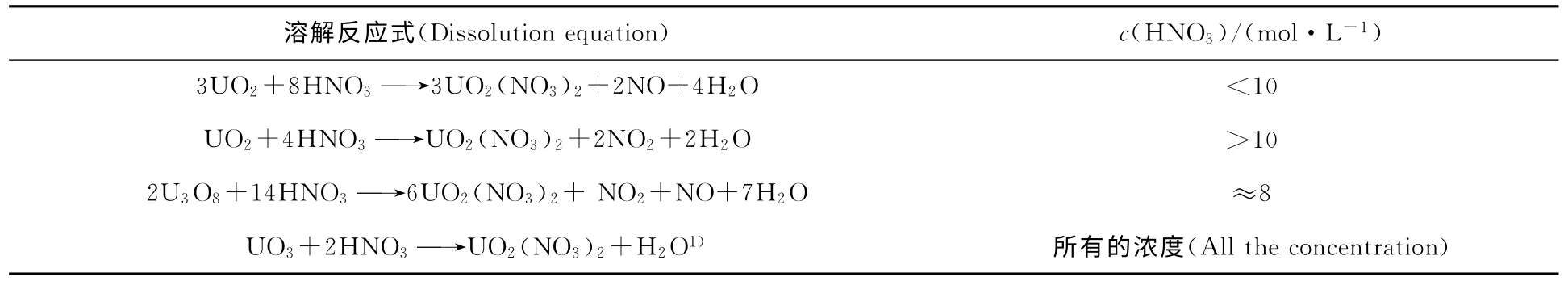
表1 不同氧化态U溶解时对HNO3需求的影响Table 1 Effect of U in different oxidation states on the demand of HNO3
乏燃料中的挥发性物质(Xe、Kr、14C等)在480℃下氧化4h释放不完全,升高温度(750℃),延长加热氧化时间(8~10h),基本上可以完全释放。
提高温度能增大半挥发性元素的释放量,按照以下次序:Ru+Rh、Tc、Cs、Te和Mo。在使用空气或O2作氧化剂时,氧化温度提高至1 250℃,能分离掉大部分半挥发性元素。
氧化挥发法由于乏燃料元件的溶解加快,并对挥发性、半挥发性元素具有很好的去除效果,降低了对后处理后续工艺的影响。但是对于Pu、Np等锕系元素、贵金属元素以及过渡金属元素,如Pd、Zr、Tc等影响如何,还需要进一步研究。
2 PUREX流程的改进
20世纪40年代后期至50年代,美国为了生产武器级Pu(WgPu)发明了军用后处理流程(PUREX),并建造运行了第一代后处理厂(军用后处理厂)。20世纪60年代,美、英、法等国将军用后处理的PUREX流程修改后用于处理核电站乏燃料元件,建造运行了第二代后处理厂。20世纪80年代,英、法为适应处理较高燃耗(40~50GWd/tU)的动力堆乏燃料,以及减少放射性物质向环境的排放,改进了PUREX流程工艺、关键设备和废物处理技术,设计、建造和运行较先进的第三代后处理厂(THORP厂、UP2-800、UP3)[6],使PUREX工艺发展成为比较成熟的后处理工业技术。但是这种只分离U和Pu的第三代后处理技术,高放废物中含有长期高放射毒性的次要锕系元素(MA:Np、Am、Cm等)、中期高释热裂变产物(90Sr和137Cs)以及长寿命裂变产物(LLFP:99Tc等),最终深地质处置所需处置库容积较大,安全监管的时间至少要1万年以上。因此,对这种所谓的标准PUREX工艺流程还必须进一步改进。
20世纪90年代后期,美国开始开发乏燃料先进湿法后处理技术,因为能源部核能办公室认为先进湿法工艺最适合处理目前轻水堆乏燃料[7]。他们的主要目标是要避免在流程的任何阶段分离纯Pu,以便减少核扩散风险。为此开发了UREX(uranium extraction)流 程[8],用30%TBP/nDD萃取分离U(99.9%)和Tc(≥95%),用乙异羟肟酸(AHA)作洗涤络合剂,以防止Pu和Np的萃取,把Pu、MA和FP留在废液中供下一步分离,采用UREX+变体流程[3]19分离超铀废物(TRU)。
2.1 德国改进型PUREX流程(IMPUREX)
为了改进PUREX流程,在共去污循环中,优化工艺参数,使用有机无盐试剂,简化流程,减少放射性废液产生量,在流程中同时分离U、Pu、Np和Tc。
德国核燃料后处理公司提出了改进型PUREX流程(IMPUREX)[9],采用了以下新工艺:
1)用硅藻土床过滤作料液第二级澄清,除去料液中细颗粒,以避免有机物夹带造成去污因素降低;
2)用电化学方法把Pu的价态调节到Pu(Ⅳ);
3)优化HA萃取柱操作条件,提高料液c(HNO3)>5mol/L和操作温度50℃,采用双酸洗涤,提高去污因子;
4)在U/Pu分离段采用电解还原脉冲柱,确保U产品有高的Pu去污因子;
5)用碳酸肼洗涤有机溶剂,电氧化法破坏碳酸肼,减少中放废液产生量。
2.2 日本先进的PUREX后处理流程(PARC)
日本核燃料公司[10]在首端维持较高酸度5~6mol/L HNO3,并延长保温(≈100℃)时间,使Pu氧化成Pu(Ⅵ),Pu(Ⅵ)可将Np氧化成Np(Ⅵ),在共去污循环中共萃取U、Pu和Np,用硝酸羟胺(HAN)还原反萃Pu+Np。英国核燃料有限公司(BNFL)与俄罗斯镭学研究所合作研究[11],采用硝酸羟胺还原反萃Pu+Np,制备MOX燃料。
为了进一步减少废物体积,把高放废物的放射毒性降低,日本原子能研究所开发了一种先进的PUREX后处理流程:PARC(partitioning conundrum key)流程,实现了U、Pu、Np、Tc的分离[12-14]。其主要的改进有:
1)在溶解器和调料槽中通入NOx气体,把Np(Ⅴ)氧化成Np(Ⅵ),并将I-氧化成I2,IO-3还原为I2;
2)在1A萃取下半段加入NH4VO3氧化剂使Np保持在Np(Ⅵ),促进U、Pu、Np、Tc共萃取;
3)在1B中U/Pu分离前先用高酸洗涤Tc;
4)用正丁醛选择性还原反萃Np,使Np与U、Pu分离后,再用异丁醛还原反萃Pu;
5)用碳酸氢丁胺洗涤有机溶剂,使其有效地净化,使用无盐试剂,可减少二次废物。
2.3 无盐试剂
在PUREX流程中,Np的价态和TBP萃取行为比较复杂,在共去污循环中要让100%Np与U、Pu共萃取(使Np保持在Np(Ⅵ))或者让Np全部留在萃残液中(使Np保持在Np(Ⅴ))是很困难的,这是因为Np的复杂的化学性质决定的,Np(Ⅴ)与U(Ⅵ)可形成阳阳络合物,从而影响到Np(Ⅴ)在TBP中的分配。提高萃取进料液的酸度和温度,可使Np的萃取率达到90%以上[15]。共萃取U、Pu和Np的有机溶剂,使用有机还原剂使Pu和Np一同还原反萃,实现与U分离:
1)乙醛肟(CH3CH N—OH)[16]
将Pu(Ⅳ)、Np(Ⅵ)还原成Pu(Ⅲ)、Np(Ⅴ),与Tc(Ⅶ)离子间无相互作用。
2)二甲基羟胺((CH3)2NOH)[17]
能很快地还原Pu(Ⅳ)、Np(Ⅵ)到Pu(Ⅲ)、Np(Ⅴ),实现Pu、Np和U的分离。
3)甲异羟肟酸(HCONHOH,FHA)和乙异羟肟酸(CH3CONHOH,AHA)[18-20]
羟肟酸能快速地将Np(Ⅵ)还原到Np(Ⅴ),而不会形成Np(Ⅳ)。+2CH3→CONHOH+2CH3COOH+N2+2H+。还原Pu(Ⅳ)的速率较慢,但能与An(Ⅳ)形成稳定的络合物,可用于Pu(Ⅳ)和Np的反萃,从U中分离出Np和Pu。在PUREX流程处理高Pu含量的燃料时,用U(Ⅳ)反萃Pu需要的U(Ⅳ)还原剂用量过大,解决的方法目前有俄罗斯在反萃装置内电解再生U(Ⅳ),英、美等使用短链异羟肟酸RCONHOH作还原/络合剂,从负载有机相中还原反萃Np和Pu。异羟肟酸在HNO3溶液中容易水解,甲异羟肟酸(FHA)比乙异羟肟酸(AHA)更容易水解,AHA在1、3、5mol/L HNO3介质中降解速率的半寿命分别为300、100、60min。水解反应产生羟胺和羧酸:
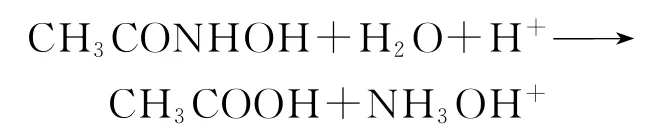
产生的羟胺将Pu(Ⅳ)还原成Pu(Ⅲ):
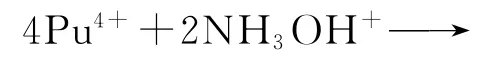
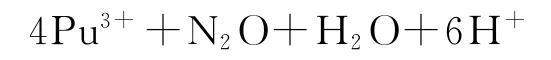
这有利于反萃Np和Pu。已建议使用0.2~0.5mol/L AHA和0.2~0.5mol/L HNO3从负载有机相中反萃Np和Pu,实现与U的分离。
4)二羟基脲(DHU)[21]
二羟基脲结构式如下:
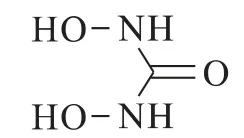
它能较快地将Pu(Ⅳ)、Np(Ⅵ)还原为Pu(Ⅲ)、Np(Ⅴ),从负载了U、Pu、Np的TBP/OK体系中定量反萃Pu和Np,与U分离。
3 后处理工艺过程涉及的几个关键基础问题
后处理工艺经过半个多世纪的发展,技术相对比较成熟。但至今涉及到一些基础科学问题没有深入研究和清楚的解释,影响到工艺运行稳定性。体现在以下几个方面。
3.1 Np的行为和走向研究
237Np是乏燃料中重要核素之一。元件在反应堆运行过程中,237Np可由235U或238U通过中子俘获反应及β衰变而得到:

在一般的轻水堆乏燃料中,237Np的生成量约为0.5~0.7kg/t乏燃料,随着反应堆燃耗的不断加深,乏燃料中的237Np含量也将不断提高。
镎在水溶液中化学形态比较复杂,在一定条件下Np以(Ⅲ)、(Ⅳ)、(Ⅴ)、(Ⅵ)、(Ⅶ)五种价态存在。在没有络合剂存在时,前四种价态都以水合离子存在。在Np的各种价态中,五价镎在溶液中最稳定,此时,镎以带一个电荷的水合镎酰离子存在,形成对称的线形键(O—Np—O)。
后处理过程中Np价态受氧化还原剂、络合剂、温度、酸度以及自身歧化等多种因素影响,其中最主要的是氧化还原、络合以及歧化的影响。在铀钚分离过程中,使用氧化还原剂可将Np(Ⅵ)还原到Np(Ⅴ),或进一步还原到Np(Ⅳ)。如采用硝酸羟胺及其衍生物如二甲基羟胺等无盐还原剂[22-25],可快速还原Np(Ⅵ)至Np(Ⅴ),而还原Np(Ⅴ)到Np(Ⅳ)的速度很慢,这样Np随Pu进入后续Pu纯化循环。采用Fe2+、U(Ⅳ)等还原剂可使Np从五价进一步还原到四价,从而使大部分Np随U一同进入铀纯化循环。还有一类兼顾络合性质的还原剂——羟肟酸类衍生物,如国际上研究较多的甲羟肟酸和乙羟肟酸等[26-27],该类还原剂可还原Np(Ⅵ)至Np(Ⅴ)、Pu(Ⅳ)至Pu(Ⅲ),同时对于四价锕系元素具有一定的络合能力。因此,该试剂可以有效地将Np和Pu一同反萃到水相。
20世纪80年代中期,Cousson等[28-31]利用X射线衍射的方法证实了Np(Ⅴ)所生成的配合物晶体中阳阳离子络合物的存在。随后,锕系元素所生成的阳阳离子键在更多的晶体结构中被发现。图1和图2是已发现的两种含Np(Ⅴ)的晶体结构模型图。从图1、2可以看出,Np(Ⅴ)通过内配位的方式相互结合形成阳阳离子络合物,并且在晶体结构中,Np(Ⅴ)离子可与更多的阳离子络合(最多4个)形成阳阳络合物。图3列出了Np(Ⅴ)在晶体结构中可能存在的8种阳阳离子络合模式。但在后处理工艺过程中,Np(Ⅴ)的阳阳络合行为研究的较少,只有鲜亮[32]开展过相关研究,实验证实Np(Ⅴ)与Np(Ⅴ)之间、Np(Ⅴ)与U(Ⅵ)之间可以形成阳阳络合离子,从而影响到Np的走向。但是Np(Ⅴ)与后处理工艺过程中其它阳离子是否形成阳阳络合物、形成络合物的稳定性以及络合物的萃取行为还不清楚。
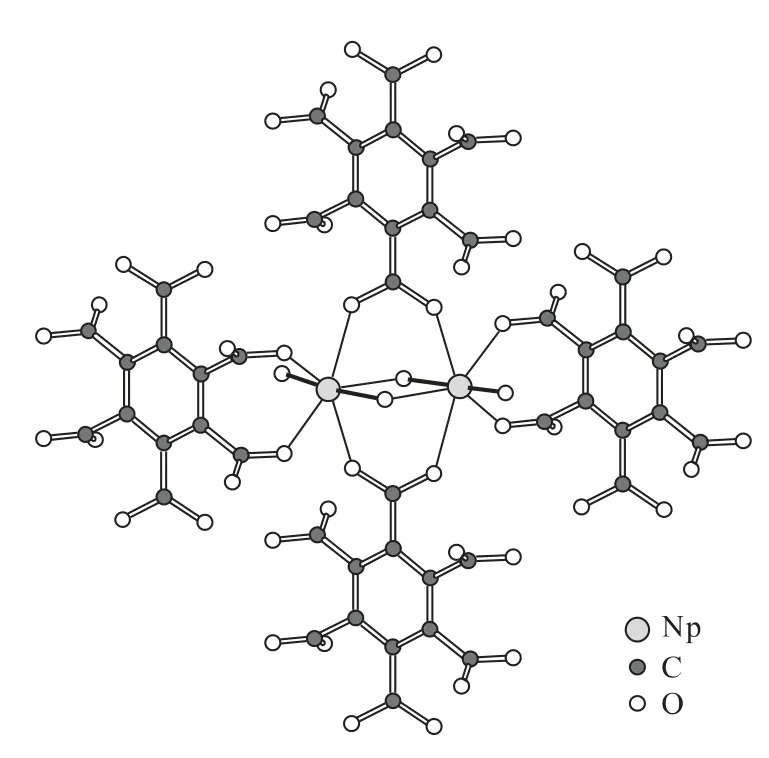
图1 Na4[(NpO2)2C6(COO)6]·8H2O晶体中的阳阳离子络合结构图Fig.1 Cation-cation complexation structure of Na4[(NpO2)2C6(COO)6]·8H2O crystal
Np(Ⅴ)除了形成阳阳离子络合物外,在水溶液中还存在不同价态间的歧化和反歧化过程[33]。近年来,部分学者通过量化计算[34]手段对An(Ⅴ)的歧化过程进行推测,认为An(Ⅴ)的歧化先要经历阳阳离子络合这一中间过程,随后,阳阳离子中间产物将通过两次质子化过程最终生成四、六价态的锕系元素(图4)。
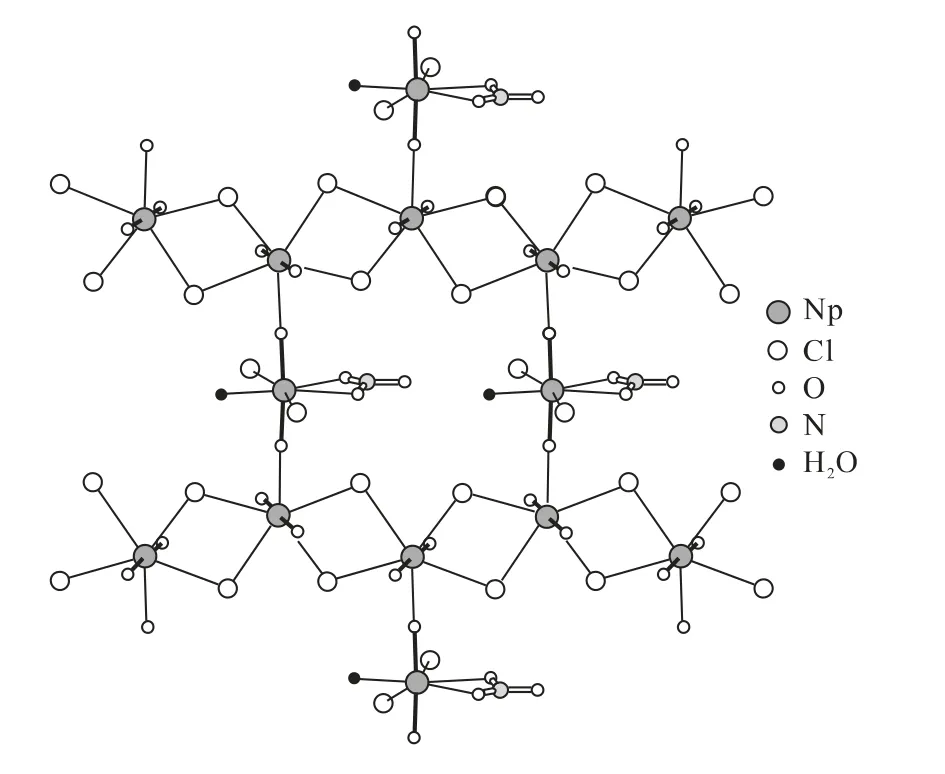
图2 Cs4[(NpO2)2Cl4NpO2Cl2(NO3)(H2O)]晶体中的阳阳离子络合结构图Fig.2 Cation-cation complexation structure of Cs4[(NpO2)2Cl4NpO2Cl2(NO3)(H2O)]crystal
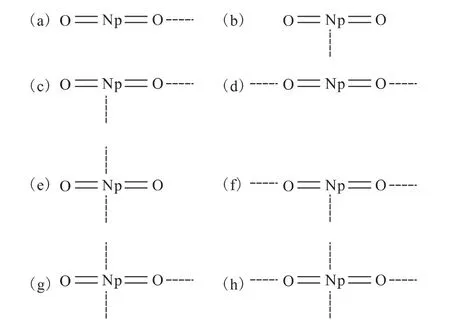
图3 晶体中可能存在的阳阳离子络合模型Fig.3 Possible cation-cation complexation model in crystal
由于上述原因,使得Np的价态控制变得十分困难,因此,Np走向相关基础问题仍是当前国际的研究热点之一。
3.2 高浓Pu的水解聚合研究
至今为止,已发现Pu的同位素共有20个,但在乏燃料后处理中,最为关注的Pu同位素是239Pu、240Pu、241Pu以 及238Pu。Pu属 于 第ⅢB族,由于其电子壳层的结构决定,不仅可以把7s轨道上电子作为价电子给出,而且也可以把5f轨道上电子作为价电子给出。因此,Pu离子在溶液中分别以Pu3+、Pu4+、和离子形式存在。因为价态之间的平衡关系以及价态之间转换的动力学,所以这4种离子形式以明显的浓度共存于同一溶液中是可能的。
关于Pu的水溶液化学研究开展的相对较多,但只是单一体系下相对定性的结果,有些研究结论也存在歧义。如Pu水合作用的一个重要特点是水解反应,水解反应程度随pH值的增大而加重。反应方程式如下:

经单一体系实验证明,不同价态Pu离子随pH值增大的水解顺序为:

Choppin等[35]提出了对Pu水合的完整描述,如图5所示,水合熵的顺序如式(3)所示:

由此可见,水解顺序与水合熵顺序不一致,差异原因是水合熵只与该阳离子基团的净正电荷有关,而水解反应是水分子与金属原子本身反应的结果。
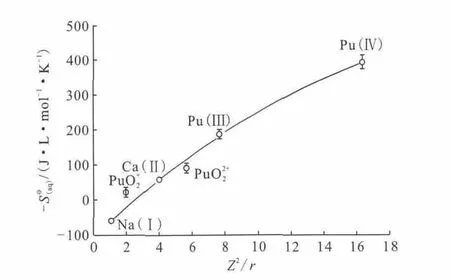
图5 水解(随pH值变化)对水合钚的阳离子基团浓度的影响[35]Fig.5 Effect of hydrolysis on the concentration of cationic group of the hydrated plutonium[35]
Toth等[36]认为Pu(Ⅳ)水解及其初级初始水解产物聚集作用的报道存在片面性。因为未适当地控制实验条件,所以某些早期数据的正确性可能遭到质疑。为了更精确地研究这一问题,对于这些实验体系应作进一步的考虑。研究人员感兴趣的主要领域仍然是Pu(Ⅳ)的水解产物与溶液中存在其它反应物之间的相互作用。这不仅对阐明新型化学相互作用有相当价值,对于充分理解这些相互作用的程度也很需要,以确保在后处理操作中对Pu有完善的控制。
综上所述,Pu的溶液化学研究还相对薄弱,特别是后处理TBP/OK-HNO3体系中Pu的基础化学研究。Pu的基础化学研究相对薄弱的关键在于以下几个方面:(1)Pu的化学毒性和辐射毒性造成研究过程的防护条件苛刻,许多实验室难以具备相关的实验防护条件;(2)Pu的化学形态测试研究条件不成熟,除了UV-Vis、拉曼、激光诱导光声光谱外,同步辐射目前不具备开展Pu化合物分析的条件;(3)致力于从事相关研究的科研队伍较少,缺乏基础研究的长远计划。中国原子能科学研究院核燃料循环后处理放化实验设施于2015年投入使用,具备开展Pu的水解聚合研究的条件。
Pu的水解聚合研究重点应围绕在后处理TBP/OK-HNO3体系中高浓Pu的水解聚合、阳阳络合物形成、歧化与价态变化规律以及对Pu的萃取行为影响研究方面。
3.3 高产额裂变产物化学形态和行为研究
在乏燃料后处理工艺研究中,关注的高产额裂变产物主要是Ru、Tc、Zr等,表2列出上述3个主要裂变产物的同位素产额值。
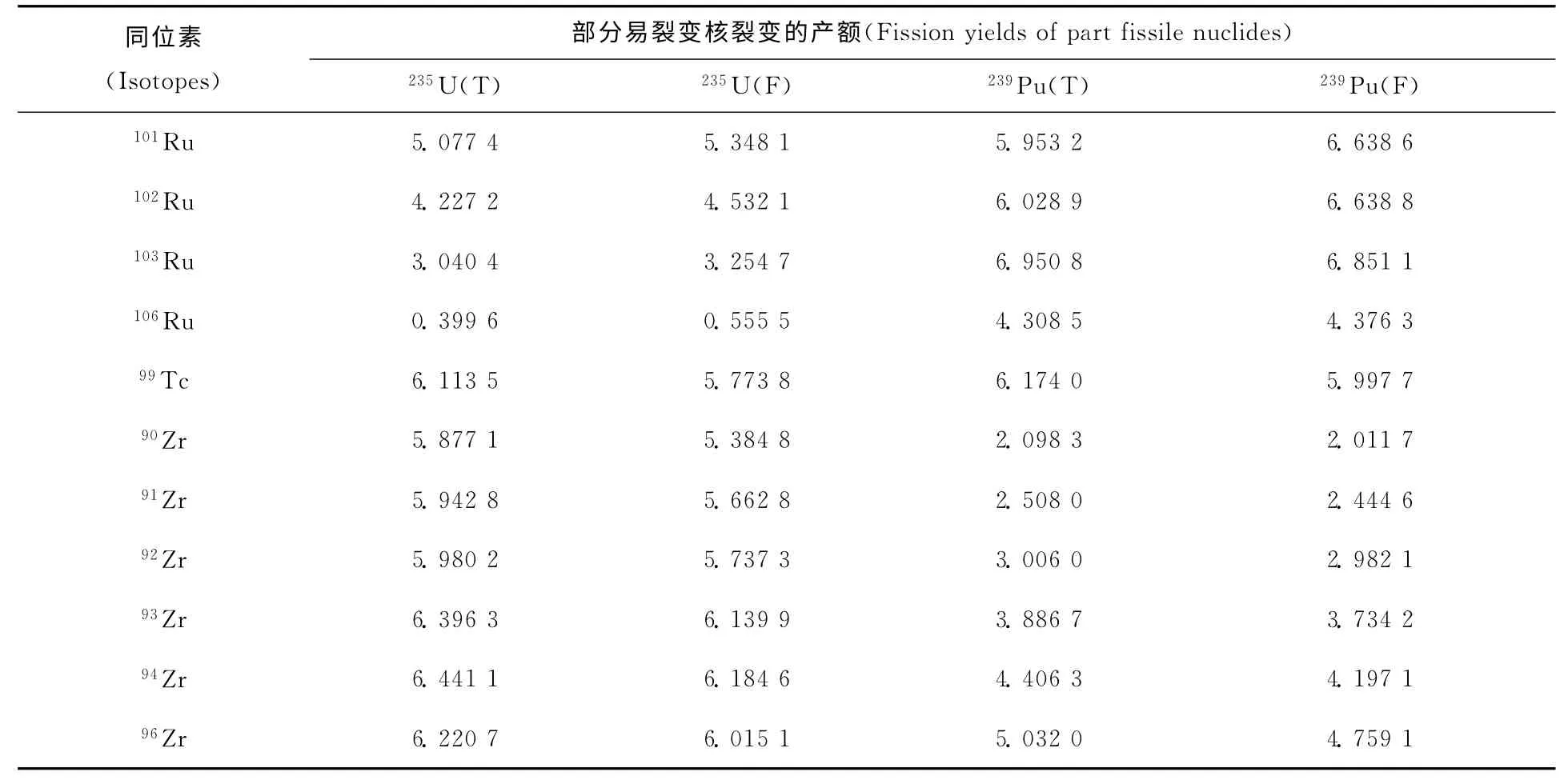
表2 主要高产额裂变产物Ru、Zr和Tc的同位素裂变产额值Table 2 Isotope fisson yields of major high-yield fisson products Ru,Zr and Tc
早期生产堆乏燃料元件燃耗浅,裂变产物Ru、Zr、Tc等裂变产物含量相对较低,它们的行为对后处理工艺的影响并不十分明显。随着动力堆燃耗的不断增加,裂变产物Ru、Zr、Tc等含量增加,从而造成Zr和Mo在溶解液中形成次级沉淀,造成溶解液不稳定,甚至钚也被共沉淀而损失;元素Tc浓度增高,对四价铀还原反萃钚有影响;Ru、Zr等含量增大在U、Pu共萃取段形成三相造成U、Pu的损失;燃耗增加,加剧了硝酸和溶剂的辐解,从而使工艺萃取端对Ru等保留增加,造成对裂变产物核素的去污较低等。因此,随着动力堆燃耗的增加,高产额裂变产物化学形态和行为研究日益引起人们的高度重视。关于高产额裂变产物元素化学性质、溶剂萃取、在后处理中走向和行为等过程,文献[37]进行了全面的论述,但是针对这几个核素在工艺过程涉及的基础问题没有进一步说明。
1)Ru
后处理是在硝酸体系中进行Np和Pu的调价,并进行U和Pu的回收提取、裂变产物的去污。在硝酸体系中由于亚硝酸钌RuNO3+键的稳定性,形成上百种钌的亚硝酰配合物,同时不同形态的亚硝酰钌硝酸根配合物之间可以相互转化(如图6),不同亚硝酸根的配合物也会相互转化,硝酸根与亚硝酸根之间相互转化。因此,在后处理工艺过程中裂变产物钌的化学形态众多,行为比较复杂。
关于Ru在硝酸体系中化学行为研究的比较深入,随着动力堆乏燃料燃耗的增加,Ru的含量也增加。因此,针对裂变产物钌还需开展以下相关基础研究:
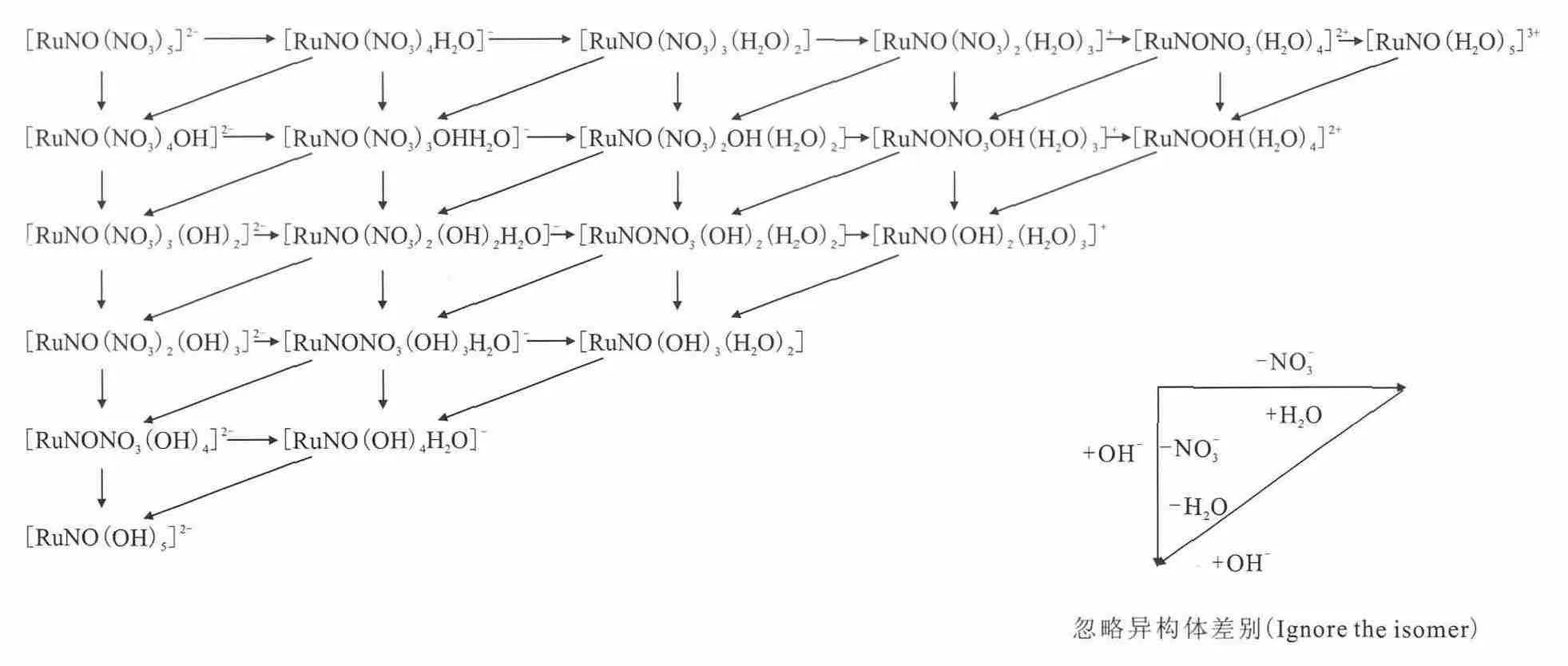
图6 单核亚硝酰钌硝酸根配合物的转化(忽略异构体差异)Fig.6 Conversion of the nitrate complex of single core ruthenium nitrosyl(ignore the isomer)
(1)后处理工艺首段高温加热除钌的机理研究;
(2)Ru的形态及其配合物相互转化及控制研究;
(3)强α辐射条件下,萃取剂TBP的降解产物对Ru的保留研究;
(4)硝酸体系下,不同形态Ru与Np(Ⅴ)、U(Ⅵ)等之间是否形成阳阳络合物等等。
2)Zr
在235U热中子诱发裂变的产物中,元素Zr的含量最大。Zr在硝酸中以硝酸锆(Zr(NO3)4·5H2O)和硝酸锆酰(ZrO(NO3)2·2H2O)形式存在。在低酸情况下,Zr很难以单一的Zr4+形式存在,主要是以很稳定的锆酰离子ZrO2+形式存在,硝酸锆酰实际上是硝酸锆的水解产物。在后处理工艺研究中,由于在硝酸中单核锆的配合物可被TBP萃取,聚合锆化合物不被TBP萃取。
裂变产物Zr的化学形态相对Ru简单,但是由于Zr在后处理工艺过程中的共萃取以及盐析效应,还有一些基础问题需要深入研究:
(1)乏燃料首端溶解液不稳定性以及次生沉淀产生机理研究;
(2)Zr在强辐射条件下溶剂中保留机理及方式研究;
(3)Zr在1A萃取器中的行为及萃取到有机相中化合物深入研究等。
3)99Tc
随着动力堆乏燃料燃耗的加深,99Tc的含量升高,由于其半衰期长,达2.11×105a,所以在乏燃料后处理回收铀的产品中对99Tc含量有限制。同时在U/Pu分离步骤若采用四价铀作为Pu的还原反萃剂时,Tc具有特殊的影响,因此Tc在PUREX流程中的走向应予以高度重视。
主要研究涉及以下几个方面:
(1)Tc与后处理工艺中的氧化还原剂作用机理及其对Pu的价态影响研究;
(2)在硝酸体系下,Tc的各种价态之间的变化规律及结构分析研究;
(3)不同价态Tc与不同价态Pu之间的氧化还原反应机理研究;
(4)TcO2+与其它阳离子是否形成阳阳络合物离子及其结构分析研究等。
4 结束语
全球经济增长和人口增加对能源的需求不断增长,以及应对全球气候变暖要求减排CO2和保护环境的压力,必将推动核电较快速地发展。
发展核电,只发展热堆和实现“一次通过”的开式循环是不可行的,不仅U资源利用极低,而且核废物处置需要的地质处置库容积太大,与生物圈隔离时间太长(几十万年以上),这是难以接受的。因此,为了核电能可持续发展,为人类持续提供安全而又清洁的能源,在发展热堆核电的同时,也要适时地发展快堆核电,实施闭式快堆燃料循环,这样不仅可充分利用U资源,减少核废物体积和长期放射毒性,使地质处置库与生物圈隔离时间缩短到1 000a以内。
热堆乏燃料后处理工艺的发展趋势是改进PUREX流程工艺,改进首端工艺,分离掉易挥发性FP,优化共去污循环参数,选用高效有机无盐还原剂,在PUREX流程中分离U、Pu、Np和Tc。
在湿法后处理工艺改进中,在考虑Np、Pu调价的同时,必须关注Np的行为和走向、高浓Pu的水解聚合、高产额裂变产物元素的化学行为和形态等方面的基础研究,才能完善湿法后处理工艺,提高后处理厂的安全运行。
致谢:在文献调研、整理、编写过程中,得到了王方定先生的精心指导和修改,在此表示衷心的感谢。也感谢丁有钱、杨金玲同志在论文编辑和文献查阅方面给予的帮助。
[1]OECD.Nuclear energy today[G].2nd Ed.OECD/NEA.Paris:NEA,2012:No6855.
[2]Peter R,Peter K.Fast reactors provide sustainable nuclear power for“thousands of years”[J/OL].Vienna:International Atomic Energy Agency,2015.http:∥www.iaea.org/newscenter/news/2013/fastreactors.html.
[3]IAEA.Spent fuel reprocessing options[R].Vienna:IAEA,2008:7-10,19.
[4]OECD/NEA.OECD/Nuclear Energy Data,advanced nuclear fuel cycles and radioactive waste management[G].Paris:NEA,2006:No 5990.
[5]Salvatores M,Geckeis H,Gompper K,et al.NSCWPFC task force on potential benefits and impacts of advanced fuel cycles with actinide partitioning and transmutation(WPFC/TFPT)[C].Japan:[s.n.],2011:397164.
[6]Fedorov Y S,Anisimov O P,Zilberman B Y,et al.Modernization of SNF WWER-440reprocessing at RPA“Mayak”[C].Japan:[s.n.],2011:3910.
[7]Kenneth L N,Gregg J L.Advanced separation techniques for nuclear fuel reprocessing and radioactive waste treatment[M].UK:Woodhead Publishing Limited,2011.
[8]昝亮彪,译.轻水堆燃料后处理方法比较[J].乏燃料管理及后处理,2009(2):17-27.
[9]Eichler R等,改进型普雷克斯流程:一种进一步降低后处理成本的途径[G]∥蒋云清主编.乏燃料后处理’94.北京:核科学技术研究所,1995:125-130.
[10]Koma Y.Recovery of minor actinides in spent fuel reprocessing based on PUREX process[C]∥RECOD.5thInternal Nuclear Conference on Recycling,Condition and Disposal,France,Oct 25-28,Paris:SFEN,1998.
[11]Taylor R J,May I,Denniss I S,et al.The development of chemical separation technology for an advanced PUREX process[C]∥RECOD.5thInternal Nuclear Conference on Recycling,Condition and Disposal,France,Oct 25-28,Paris:SFEN,1998.
[12]Uchiyama G.一种改进Purex流程(PARC流程)的开发[G]∥蒋云清主编.乏燃料后处理’98.北京:核科学技术研究所,2000:94-98.
[13]Uchiyama G.PARC流程中U、Np、Pu和Tc的萃取行为[G]∥蒋云清主编.乏燃料后处理’98.北京:核科学技术研究所,2000:123-128.
[14]Asakusa T,Uchiyama G,Kihara T,et al.A test line newly installed in NUCEF and research program on advanced reprocessing process by utilizing it[C]∥RECOD.5thInternal Nuclear Conference on Recycling,Condition and Disposal,France,Oct 25-28,Paris:SFEN,1998.
[15]Brikett J E,Carrot M J,Fox O D,et al.Controlling neptunium and plutonium within single cycle solvent extraction flowsheet for advanced fuel cycle[J].J Nucl Sci Technol,2007,44(3):337-347.
[16]Koltunov V S,Taylor R J,Baranov S M,et al.Acetaldoximiea promising reducing agent for Pu and Np ions in the PUREX process[C]∥Atalanta’2000,France,Oct 24-27,France:[s.n.],2000.
[17]Koltunov V S,Baranov S M.Organic derivatives of hydrazing and hydroxylamine in future technology of spent nuclear fuel reprocessing[C]∥International Conference on Evaluation of Emerging Nuclear Fuel Cycle Systems,France:[s.n.],1995.
[18]Taylor R J,May I,Wallwork A L,et al.The application of fermo-and aceto-hydroxamic acid in nuclear fuel reprocessing[J].J Alloys Compds,1998,271-273:534-537.
[19]May I,Taylor R J,Brown G.The formation of hydrophilic Np(Ⅵ)complexes and their potential application in nuclear fuel reprocessing[J].J Alloys Compds,1998,271-273:650-653.
[20]Taylor R J,Denniss I S,May I.Hydroxamic acidsnovel reagents for advanced PUREX process[C]∥Atalanta’2004,France,Oct 24-27,France:[s.n.],2004.
[21]YAN T H,Zheng W F,Ye G A,et al.The route of Np in the U/Pu separation using dihydroxyure as reductant[C]∥Global 2011,Japan:[s.n.],2011.
[22]张先业,叶国安,肖松涛,等.单甲基肼还原Np(Ⅵ)Ⅰ:反应动力学研究[J].原子能科学技术,1997,31(3):193-198.
[23]张先业,叶国安,肖松涛,等.单甲基肼还原Np(Ⅵ)Ⅱ:PUREX流程中U-Np分离的研究[J].原子能科学技术,1997,31(4):315-320.
[24]何辉,胡景炘,张先业,等.N,N-二甲基羟胺对Pu(Ⅳ)的还原反萃和相应的计算机模型[J].核化学与放射化 学,2001,23(2):65-71.
[25]李小该,叶国安,何辉,等.APOR流程1B槽中镎的走向行为研究[J].原子能科学技术,2010,44(2):129-137.
[26]郑卫芳.乙异羟肟酸在先进后处理流程中的应用研究[D].北京:中国原子能科学研究院,2007.
[27]Brikett J E,Carrott M J,Fox O D,et al.Plutonium and neptunium stripping in single cycle solvent extraction flowsheets:recent progress in flowsheet testing[C]∥Atalanta’2004,France,Oct 24-27,France:[s.n.],2004.
[28]Cousson A,Dabos S,Abazli H,et al.In abstracts of the 1st international conference on chemistry and technology of lanthanides and actinides[C].Venice:[s.n.],1983.
[29]Cousson A,Dabos S,Abazli H,et al.Crystal structure of a neptunylcation-cation complex(NpO2+)with mellitic acid:Na4(NpO2)2Cl12O12·8H2O[J].J Less-Common Met,1984,99(2):233-240.
[30]Cousson A.Structure du bis(benzenetetracarboxylate-1,2,4,5)de pentaneptunyle(Ⅴ)et de triammonium a seprt molecules deau[J].Acta Crystallogr,Sect C,1985,41:1758-1761.
[31]Tomilin S V,Volkov Y F,Melkaya R F,et al.Crystal structure of the aquachloro complex of neptunium(Ⅴ)with cesium CsNpO2Cl2(H2O)[J].Radiokhimiya,1986,28:304.
[32]鲜亮.Np(Ⅴ)阳阳离子络合行为及其对Np在后处理流程中的走向影响初步研究[D].北京:中国原子能科学研究院,2013.
[33]Hindman J C,Sullivan J C,Cohen D.Kinetics of the neptunium(Ⅲ)-neptunium(Ⅴ)reaction in perchlorate solution[J].J Am Chem Soc,1958,80(8):1812-1814.
[34]Steele H,Taylor R J.A theoretical study of the inner-sphere disproportionation reaction mechanism of the pentavalent actinyl ions[J].J Inorg Chem,2007,46(16):6311-6318.
[35]Choppin G R.钚的溶液化学[M]∥胡晓丹,丁戈龙,刘文彬.钚化学.北京:原子能出版社,2010:121-130.
[36]Toth L M,Friendman H A,Osborne M M.Pu(Ⅳ)的水合聚合物化学[M]∥胡晓丹,丁戈龙,刘文彬.钚化学.北京:原子能出版社,2010:131-136.
[37]林灿生.裂变产物元素过程化学[M].北京:原子能出版社,2012:2-405.
