Bi2Se3、Bi2Te3和Sb2Te3的声子和热力学性质的第一性原理研究
2015-12-17刘以良
周 彤,段 婕,张 莉,刘以良
(1.四川大学原子与分子物理研究所,四川 成都 610065;2.西南民族大学电气信息工程学院,四川 成都 610041)
Bi2Se3、Bi2Te3和Sb2Te3的声子和
热力学性质的第一性原理研究
周 彤1,段 婕1,张 莉1,刘以良2
(1.四川大学原子与分子物理研究所,四川 成都 610065;2.西南民族大学电气信息工程学院,四川 成都 610041)
基于第一性原理的密度泛函理论,在未考虑和考虑自旋-轨道耦合(SOC)的情况下分别优化拓扑绝缘体Bi2Se3、Bi2Te3和Sb2Te3的结构,计算它们的声子谱及热力学性质.基于广义梯度交换相关泛函及SOC效应,计算得到三种物质的声子频率比不考虑SOC时更吻合实验数据.最后计算出三种物质的赫尔姆赫兹自由能F,内能E,等体热容CV和熵S随温度的变化趋势.
第一性原理;声子谱;热力学性质;自旋-轨道耦合
Bi2Se3、Bi2Te3和Sb2Te3是一种层状材料,它们具有优异的热电性能的同时还有奇特的拓扑绝缘特性. Bi2Se3、Bi2Te3和Sb2Te3材料有着越来越重要的潜力,主要原因是它有着寿命长、无温室效应、无噪音、稳定性高等优点[1].材料的结构、声子谱,以及热力学性质对理解和分析材料的基本性能是非常必要的.所以,计算这三种材料的声子谱以及热力学性质对更深入地理解和分析它们的热电性能是具有实际意义的.
本文中,在未考虑和考虑自旋-轨道耦合(SOC)的两种情况下,分别优化Bi2Se3、Bi2Te3和Sb2Te3的原胞结构,得到最稳定的结构.然后用这些稳定结构构造出2×2×2的超胞结构,利用超胞计算声子谱和声子态密度.基于计算出的声子色散关系,由简谐近似计算出热力学性质,包括赫尔姆赫兹自由能F、内能E、熵S、等体热容CV,同时讨论了声子对这些热力学性质的影响,并且将计算结果与已有的实验数据进行比较.
1 计算方法
本文所有的计算采用VASP[2]程序结合Phonopy软件[3]完成的,结构优化时用含有5个原子的原胞,Brillouin区采用10×10×10型的K点网格,平面截断能为400eV.基于密度泛函理论(DFT)在广义梯度近似(GGA)下,采用PBE[4]形式处理交换关联能,Bi、Se、Sb和Te原子的价电子分别为6s26p3、4s24p4、5s25p4和5s25p3,其都采用投影缀加平面波(PAW)[5]赝势.我们通过线性响应方法[6],计算物质的声子谱,并进一步预测了热力学性质.计算结构是优化的原胞构造的2×2×2超胞,展开平面波的截断能仍选用为400eV,K点采2×2×2的Mohkhorst-Pack类型的K点,能量收敛标准为10-5eV.
2 结果和讨论
2.1 结构优化
Bi2Se3、Bi2Te3和Sb2Te3优化后的结构参数和内坐标参数如表1所示,计算结果表明优化结构参数和实验值[7]以及其他理论计算值[8,9]都符合得很好.结果显示考虑和不考虑SOC优化结构得到的晶格常数差别不是很明显,但考虑了SOC后的晶格常数更接近实验值.
2.2 声子谱
计算得到的Bi2Se3、Bi2Te3和Sb2Te3的声子谱和声子态密度分别如图1到图3所示,Bi2Se3、Bi2Te3和Sb2Te3原胞中有5个原子,所以声散关系图中共有十五支振动模式,其声子谱中有三个声学支,十二个光学支.Bi2Se3、Bi2Te3和Sb2Te3的空间群都为D53d(R-3m),其中,Bi2Se3中Bi原子位于2c格位,Se1原子位于2c格位,Se2原子位于1a格位;Bi2Te3中Bi原子位于2c格位,Te1原子位于2c格位,Te2原子位于1a格位;Sb2Te3中Sb原子位于2c格位,Te1原子位于2c格位,Te2原子位于1a格位.由群论可知,Bi2Se3、Bi2Te3和Sb2Te3的布里渊区中心声子模的不可约表示是Г=2A1g+3A2u+2Eg+3Eu,光学模由2A1g、2Eg、2A2u和2Eu构成,其中,2A1g、2Eg是拉曼活性振动模,2A2u和2Eu是红外活性振动模.声学模由A2u模和Eu模构成.对于拉曼活性模,Eg描述了原子振动平面的剪切模,A1g描述了垂直于原子振动平面的呼吸模.
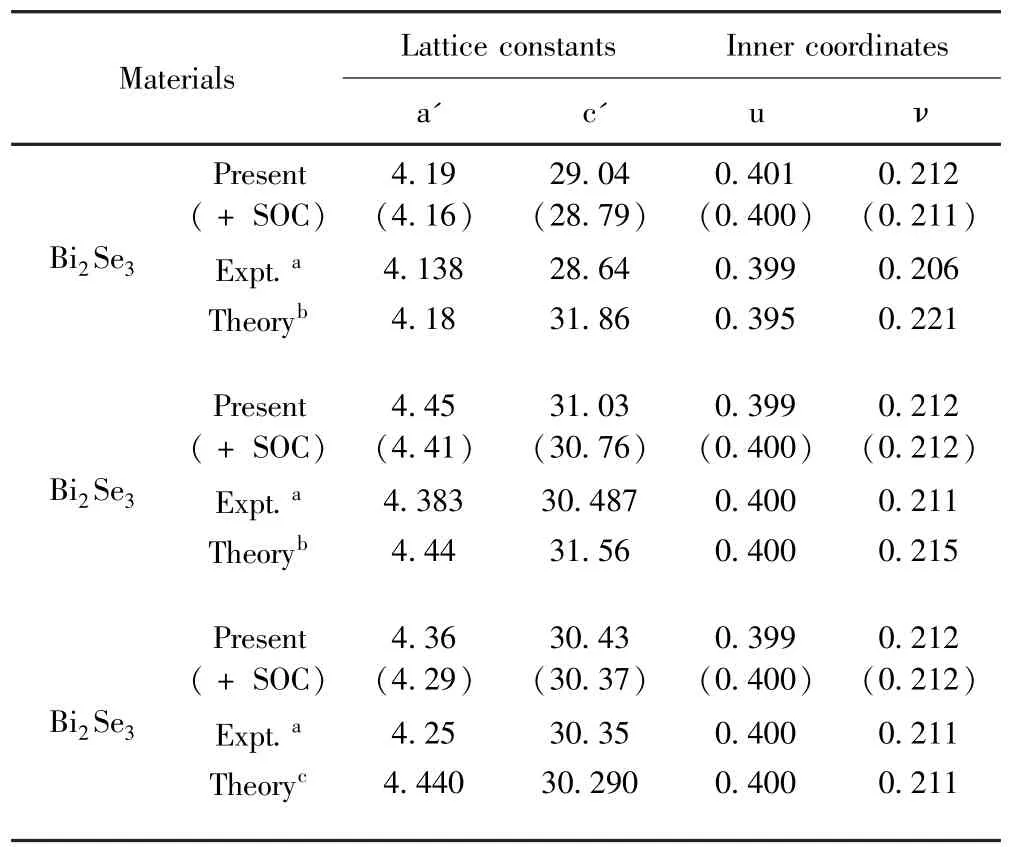
表1 Bi2Se3、Bi2Te3和Sb2Te3优化的晶格常数(便于比较,其他计算值和实验值也列在表中)Table 1 The experimental and theoretical lattice parameters and inner coordinates of rhombohedral unit cell Bi2Se3,Bi2Te3and Sb2Te3.Present,expt.and theory denote our results(the values in brackets is computed with SOC)and the values that are obtained from experiments and theoretical calculations
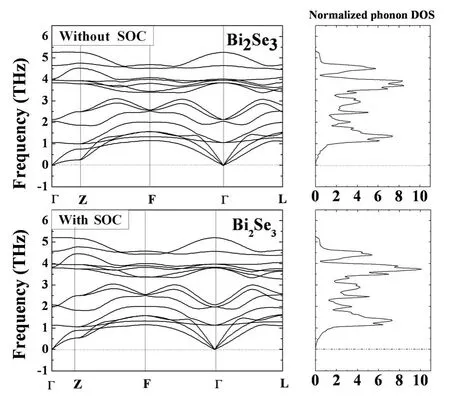
图1 Bi2Se3未考虑和考虑SOC优化结构计算得到的声子谱和声子态密度Fig.1 Calculated phonon dispersion curves and DOS for Bi2Se3optimized structures without and with SOC

图2 Bi2Te3未考虑和考虑SOC优化结构计算得到的声子谱和声子态密度Fig.2 Calculated phonon dispersion curves and DOS for Bi2Te3optimized structures without and with SOC.
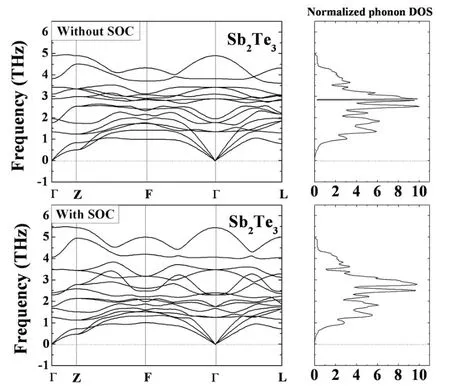
图3 Sb2Te3未考虑和考虑SOC优化结构计算得到的声子谱曲线和声子态密度Fig.3 Calculated phonon dispersion curves and DOS for Sb2Te3optimized structures without and with SOC
由计算得到Bi2Se3、Bi2Te3和Sb2Te3的声子色散曲线和对应的声子态密度可以看出,这三种物质的声子曲线和声子态密度有着很大的相似之处,光学支和声学支之间不存在间隙,是因为Bi、Se、Te、Sb原子的质量相差不大.但是由于Bi2Se3、Bi2Te3和Sb2Te3存在不同的弹性力度和离子度,这些曲线还是有一些差别.以Bi2Se3为例,其声子色散曲线中,沿着高对称方向的三条曲线特别的平滑,所以导致对应的声子态密度有3个很明显的峰.从图1,图2,图3可以看出,用未考虑和考虑SOC优化后的结构计算得到的声子谱都没有虚频,说明其结构是稳定的,而且其声子谱和声子态密度图没有很大的差别,比较相似.所以为了进一步研究,将Bi2Se3、Bi2Te3和Sb2Te3在Г点的声子频率列在表2中进行比较.表2说明未考虑SOC优化后的结构计算的声子频率比其他理论计算值[10]更接近实验值[11-12],其中Bi2Se3和Bi2Te3的其他理论计算值的最大误差分别是29.4%和21.9%,而我们计算的频率的最大误差分别是6.4%和11%.本文Bi2Se3和Bi2Te3的声子频率是用PBE计算的,与之比较的理论计算值是用LDA[10]计算的,这表明PBE比LDA能更好地处理交换关联泛函能.
比较用未考虑和考虑SOC优化的结构计算得到的声子频率,发现Bi2Se3和Bi2Te3考虑SOC优化的结构计算的声子频率低于未考虑时的值,且更接近于实验值[11-12],其误差分别为5.5%和10.1%.对于Sb2Te3的声子频率,除了EΠg,AΠ1g和AΠ2u,考虑SOC优化的结构计算的声子频率比未考虑时值略高之外,其余的也都符合上述规律.同时,本文计算的部分声子特征频率,能为其他实验以及理论研究提供一些数据基础.

表2 Bi2Se3、Bi2Te3和Sb2Te3在Г点的声子频率(单位为THz)(便于比较,其他计算值和实验值也列在表中)Table 2 Phonon frequencies at theГ point for Bi2Se3,Bi2Te3and Sb2Te3in unit of THz (For comparison,other calculated data and experimental values are also listed)
2.3 热力学性质
考虑和不考虑SOC优化,计算得到的热力学函数随温度的变化趋势相同,故都用图4来描述.

图4 Bi2Se3、Bi2Te3和Sb2Te3计算的自由能F(图a)、内能E(图b)、热容CV(图c)和熵S(图d)随温度的变化Fig.4 Calculated free energyF(Fig.a),internal energy E(Fig.b),specific heat at constant volume CV(Fig.c) and entropy S(Fig.c)variation with temperature for Bi2Se3,Bi2Te3and Sb2Te3
2.3.1 内能和赫尔姆赫兹自由能
如图4,我们给出赫尔姆赫兹自由能F、内能E、熵S和等体热容CV随温度T的变化曲线,其温度范围为0-1000K.F和E在0K是表现为零点运动,未考虑SOC优化结构得到的Bi2Se3、Bi2Te3和Sb2Te3的零点能分别为8.576 KJ/mol,6.909 KJ/mol和7.474 KJ/mol,考虑SOC优化结构得到的零点能分别8.480 KJ/mol,6.708 KJ/mol和7.405 KJ/mol,对Bi2Se3,与其他两个物质相比,零点运动对热力学函数的贡献更为重要.这三种物质中温度对Bi2Se3的F和E的影响要比其他两个更加明显,图4局部放大图可以清晰地展现这一点,主要原因是Bi2Se3有更高的平均声子频率和更低的熵值.而且,考虑SOC优化结构得到的零点能要比不考虑时都略低.
2.3.2 比热
图4(c)是未考虑和考虑SOC优化结构得到的等体热容CV随温度T的变化关系,从图中可以看出,在0-1000K的温度范围内,Bi2Se3、Bi2Te3和Sb2Te3的CV随温度T的升高而升高,到某一温度不再变化而趋于一个特定值,三个物质中,Bi2Se3的CV曲线要比其他两条低,这是因为Bi2Se3的声子态密度较小.
2.3.3 熵和焓
熵和焓是两个重要热力学参量,图4(d)显示的是未考虑和考虑SOC优化结构得到的熵值S随温度T的变化趋势,从图中可以看出,在0-1000K的温度范围内,在同一的温度下,Bi2Te3、Sb2Te3、Bi2Se3的S依次降低.
以298K时的焓H298为标准,我们用公式H-H298=E-E298
[13]计算了不同温度状态下的H-H298的值.表3,4,5是我们计算的三种物质的熵S和H-H298的结果,并与实验数据[14]进行比较.计算时,我们选定了温度范围(298.15K-熔点[15]),对Bi2Se3温度T的变化从298.15K-980K(Bi2Te3:298.15-860K;Sb2Te3:298.15-900K).以298K为标准,温度间隔为100K,我们可以得到一组H-H298的值.在低温的时候,原子主要在平衡结构附近做近视的简谐振动,因此所得结果与实验值[14]符合得很好,当然,随着温度的升高,由于非简谐效应的加剧,简谐近似下的计算结果与实验值有一定偏差.

表3 Bi2Se3未考虑和考虑SOC优化计算得到的在不同温度下的热力学函数值S和H-H298(单位:J/K/mol) (其他计算值和实验值列在表中进行比较)Table 3 The thermodynamic function valueS(J/K/mol)and H-H298(J/K/mol)of different temperature for Bi2Se3optimized structures with and without SOC

表4 Bi2Te3未考虑和考虑SOC优化计算得到的在不同温度下的热力学函数值S和H-H298(单位:J/K/mol)(实验值列在表中进行比较)Table 4 The thermodynamic function valueS(J/K/mol)and H-H298(J/K/mol)of different temperature for Bi2Te3optimized structures with and without SOC
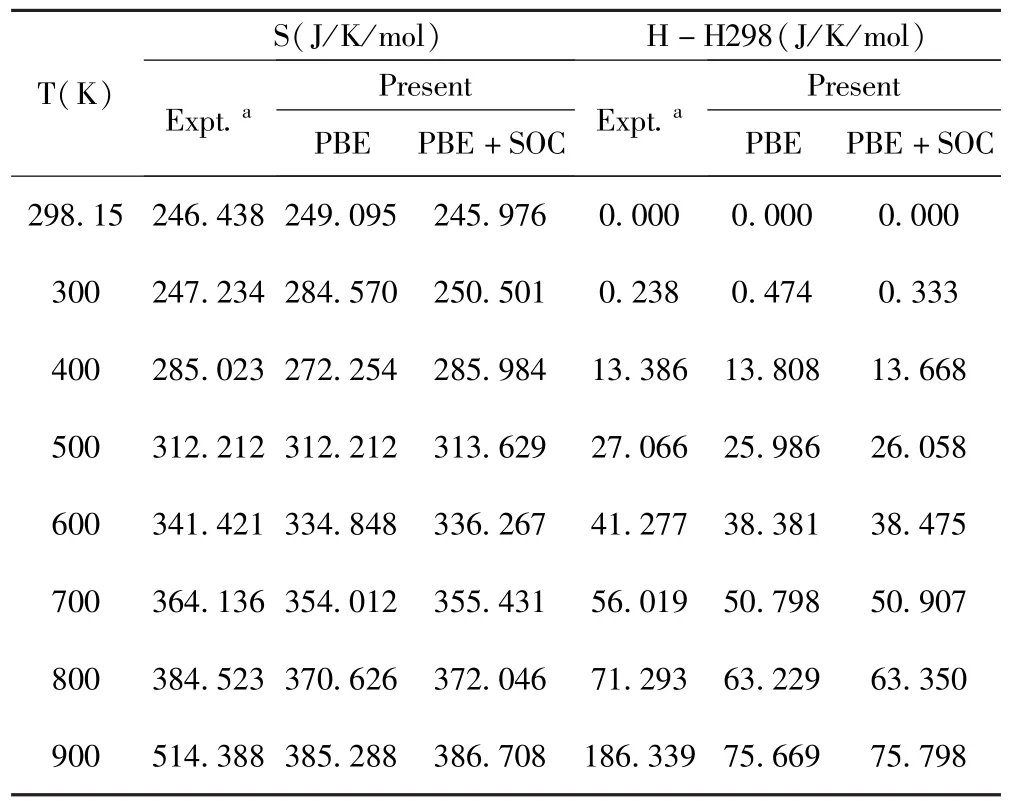
表5 Sb2Te3未考虑和考虑SOC优化计算得到的在不同温度下的热力学函数值S和H-H298(单位:J/K/mol)Table 5 The thermodynamic function valueS(J/K/mol)and H-H298(J/K/mol)of different temperature for Sb2Te3optimized structures with and without SOC
3 结论
基于密度泛函理论,计算Bi2Se3、Bi2Te3和Sb2Te3的声子谱以及热力学性质.计算结果表明PBE比LDA能更好地处理交换相关泛函.计算说明自旋-轨道耦合(SOC)对晶格常数的影响比较小,但用这个结构计算得到的声子频率比不考虑时低,而且更接近于实验值.Bi2Se3的零点运动能比Bi2Te3和Sb2Te3的大,温度对Bi2Se3的F和E的影响要比Bi2Te3和Sb2Te3更加明显.Bi2Se3的质量和晶格常数比Bi2Te3和Sb2Te3的小,这也许是同样的温度下Bi2Se3的熵值比其他两个物质的熵值小的原因之一.
[1]WANG GUO FENG,CAGIN T.Electronic structures of the thermoelectric materials Bi2Te3 and Sb2Te3 from first-principles calculations [J].Physical Review B,2007,76:075201.
[2]KRESSE G,HAFNER J.Ab initio molecular dynamics for open-shell transition metals[J].Physical Review B,1993,48:13115-13120.
[3]TOGO A.Phonopy[EB/OL].(2009-7-30)[2015-4-10]http:// phonopy.sourceforge.net.
[4]KRESSE G,HAFNER J.Ab initio molecular dynamics for open-shell transition metals[J].Physical Review B,1993,48:13115-13120.
[5]KRESSE G,JOUBERT D.From ultrasoft pseudopotentials to the projector augmented-wave method[J].Physical Review B,1999,59:1758-1761.
[6]GIANNOZZI P,GIRONCOLI S D.Ab initio calculations of phonon dispersions in semiconductors[J].Physical Review B,1991,43:7231-42.
[7]WYCKOFF R W G.Crystal Structure Descriptions[M].2nd edition,New York,1964:25-38.
[8]LIU CHAOXING,QI XIAOLIANG,ZHANG SHOUCHENG,et al.Topological insulators in Bi2Te3,Bi2Se3and Sb2Te3with Dirac cone on the surface[J].Nature Physics,2009,5:438-442.
[9]XIN LUO,SULLIVAN M B,QUEK S Y.First principles investigations of the atomic,electronic and thermoelectric of equilibrium and strained Bi2Se3&Bi2Te3with van der waals interactions[J].Physical Review B,2012,86:184111.
[10]WANG BAO TIAN,ZHANG PING.Phono spectrum and bonding properties of Bi2Se3:Role of strong spin-orbit interaction[J].Applied Physics Letters,2012,100:082109.
[11]GNEZDILOV V,PASHKEVICH Y G,BERGER H et al.Giant quantized plateau in the THz Faraday angle in the gated Bi2Se3[J].Physical Review B,2010,84:125110.
[12]SHAHIL K M F,HOSSAIN M Z,TEWELDEBRHAN D,et al.Crystal symmetry breaking in few-quintuple Bi2Te3films:Applications in nanometrology of topological insulators[J].Applied Physics Letters,2010,96:153103.
[13]MA SHENGGUI,SHEN YANHONG,GAO TAO,et al.First-principle calculation of the structural,electronic,dynamical and thermodynamic properties of γ-LiAlO2[J].International journal of hydrogen energy,2015,40:3762-3770.
[14]SEMENKOVICH S A,MELEKH B T.Thermodynamic properties of Bi2Te3,Bi2Se3and Sb2Te3[J].Chemical Bonds in Solids,1972,11: 159-162.
[15]WIKIPEDIA.Bismuth telluride[EB/OL].(2012-5-10)[2015-4 -10].http://en.wikipedia.org/wiki/Main_Page.
(责任编辑:张阳,付强,李建忠,罗敏;英文编辑:周序林)
First-principles calculations of phonon spectrum and thermodynamic properties of Bi2Se3,Bi2Te3and Sb2Te3
ZHOU Tong1,DUAN Jie1,ZHANG Li1,LIU Yi-liang2
(1.Institute of Atomic and Molecular Physics,Sichuan University,Chengdu 610065,P.R.C.;
2.School of Electrical and Information Engineering,Southwest University for Nationalities,Chengdu 610041,P.R.C.)
The structures of Bi2Se3,Bi2Te3and Sb2Te3were optimized using density-functional theory(DFT)based on first-principles with and without spin-orbit coupling(SOC)effect.Phonon spectrums and thermodynamic properties of Bi2Se3,Bi2Te3and Sb2Te3were calculated.The phonon spectrums for the three compounds were performed within the generalized gradient approximation(GGA)including SOC effects.The results showed that the calculated phonon frequencies by means of optimal structures with SOC were in better agreement with experimental data than that without SOC.Finally,the variation of their free energy(F),internal energy(E),specific heat at constant volume(CV)and entropy(S)with different temperature were calculated.
first-principles;phonon spectrum;thermodynamic property;spin-orbit coupling
O52
A
2095-4271(2015)04-0443-06
10.11920/xnmdzk.2015.04.009
2015-05-13
周彤(1989-),女,汉族,甘肃西和人,硕士研究生,主要从事凝聚态物理学的研究,E-mail:zhoutong926@163.com.
张莉(1970-),女,四川成都人,博士研究生,研究员,主要从事原子分子物理学的研究,E-mail:lizhang@scu.edu.com.
国家自然科学基金(11474209)
