鬼臼毒素C-4β位N连接衍生物的研究进展
2015-12-08张志华
张志华
(辽宁工业大学化学与环境工程学院,辽宁锦州121001)
鬼臼毒素C-4β位N连接衍生物的研究进展
张志华
(辽宁工业大学化学与环境工程学院,辽宁锦州121001)
以近年来国内外相关报道文献为基础,综述鬼臼毒素的C-4β位N连接衍生物的作用机制、合成方法、修饰手段,为该类化合物的设计和合成提供理论依据。
鬼臼毒素;C-4β位;N连接衍生物;机制;合成;修饰
鬼臼毒素(podophyllotoxin,1)是芳基四氢化萘类木脂素,可从鬼臼类植物Podophyllum peltatum L、P.emodi W、P.pleianthum H[1]及柏松属植物Callitrus drurnrnondii[2]中分离得到,对多种肿瘤具有抑制作用,但因其毒性较大,水溶性差,限制了在临床上的应用,一般制成酊剂和膏剂用于尖锐湿疣等皮肤病的治疗。为增加鬼臼毒素在临床上的应用价值,人们围绕着药物水溶性提高、细胞毒性降低以及耐药性肿瘤治疗等方面开展了大量研究工作。目前,环A~E及木脂体的骨架结构均被成功修饰过[3-7],其中最成功的例子就是在C-4位进行了糖基化修饰的依托泊苷(etoposide,2)、替尼泊苷(teniposide,3)以及其水溶性前药 (etopophos,4),它们在临床上均可用于治疗小细胞肺癌等多种恶性肿瘤[8-9]。
鬼臼毒素及其衍生物的构效关系研究显示,其分子中由3~4个环组成的稠环平面刚性结构、反式内酯环、E环中的C-4'酚羟基为抗肿瘤活性的基本部位[10-11],其中C-4β位是母核结构上一个很关键的手性部分,许多化学反应都易在此部位发生并影响构型,该位置碳氧为β-构型,若变为α-构型则抗肿瘤活性显著降低[12]。此外,化合物2中4β位上的糖基是非必要基团,除去它不影响 (结合)药物的活性和专一性[13]。而且,体外抗肿瘤筛选实验表明,N-取代衍生物的活性普遍高于其它链接原子 (S、O、Se)取代[14-15],如化合物NPF(5)、GL-331(6),其中6已进入临床Ⅱ期研究[16]。
本文将对近年来鬼臼毒素C-4β位N连接衍生物的作用机制、合成方法、修饰手段等方面进行了总结。
1 C-4β位N连接鬼臼毒素衍生物的抗肿瘤作用机制
1.1 作用于拓扑异构酶Ⅱ 拓扑异构酶控制着DNA结构的变化,能将磷酸二酯键打开或使其形成,从而控制DNA的拓扑状态。同时,拓扑异构酶II催化DNA双链的打开,在DNA复制、转录、染色体的浓缩过程中起着关键作用[17-18]。C-4β位N连接的鬼臼毒素 (如二芳基取代脲类衍生物[4])能结合拓扑异构酶II及DNA,从而形成稳定的三元复合物,阻止酶和DNA的分离,使DNA难以在切口处重新连接,最终引起细胞形态上的变化和凋亡。Xiao等[19]采用kDNA解链法发现,当C-4β-N-三羟甲基甲胺取代的鬼臼毒素浓度为10μM时,能明显降低拓扑异构酶对DNA的松弛活性。
1.2 作用于微管蛋白 微管主要由α微管蛋白 (α-tubulin)和β微管蛋白 (β-tubulin)聚合而成,它是真核细胞骨架的重要组成,与细胞的形态、运动性、细胞内外物质运输有关[11,20]。此外,它还在细胞有丝分裂过程中起着极其重要的作用,在细胞有丝分裂前期,微管聚合成为纺锤体结构,牵引染色体分别向细胞两极移动,进入到两个子细胞中,完成细胞增殖[21]。鬼臼毒素类衍生物能结合到微管蛋白的秋水仙素结合位点而导致其变形,之后进一步破坏其组装和解聚之间的动态平衡,诱导细胞的中期分裂(G2/M stage)静息。Liu等[22]报道,合成的铂和鬼臼结合的化合物 (7和8)能通过抑制微管蛋白形成而导致细胞的凋亡,并且具有裂解DNA的作用。Liu等[23]将微管蛋白用罗丹红染色后,明显观察到在C-4β位N连接鬼臼毒素衍生物 (9)的作用下,微管由细长的形态变为短粗 (化合物1~9的具体结构见图1)。
2 C-4β位N连接的鬼臼毒素的合成方法
以鬼臼毒素为原料,可直接得到氨基取代衍生物,或先合成 C-4β-氨基鬼臼毒素,再进一步与酰氯[4-6,13-14,24]、羧酸[22,25]、 异硫氰酸盐[16]、 醛[14,26]等反应, 合成的主要方法如下。
2.1 鬼臼毒素与叠氮化钠作用,经叠氮化、叠氮还原立体选择性的转化反应,得到氨基产物[27-29],或在Cu2SO4· 5H2O和抗坏血酸钠催化下,与炔丙醇生成4β-1,2,3-苯三唑鬼臼毒素类衍生物[23],反应过程见图2。
2.2 鬼臼毒素在60%的甲磺酸盐和三氧化铝作用下,与氯乙腈经Ritter反应,立体选择性地生成4β-氯酰氨基鬼臼毒素,然后用硫脲和乙酸处理,再经酰胺键水解,得到 氨基产物[16],反应过程见图3。
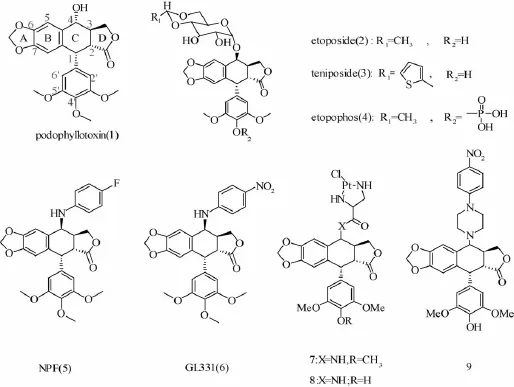
图1 化合物1~9的具体结构
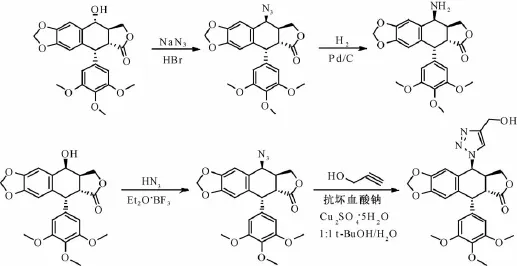
图2 具体反应过程(2.1)
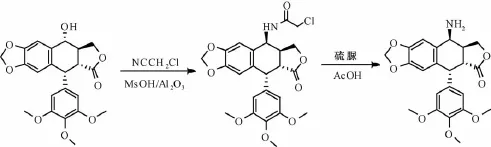
图3 具体反应过程(2.2)
2.3 鬼臼毒素在NaI或NaBr作用下,立体选择性地转化为卤化物,再经一系列胺类化合物的亲核取代反应,得到最终产物[30-31],反应过程见图4。
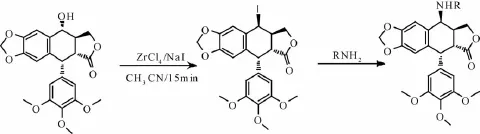
图4 具体反应过程(2.3)
2.4 Tang等[32]报道,用微生物Alternaria alternata S-f6将 4'-去甲基鬼臼苦酮与川芎嗪直接发生转氨反应,得到4-TMP-DMEP,反应过程见图5。 MCF-7细胞的作用均高于鬼臼毒素和芪类化合物,说明两者产生了协同效果[25]。
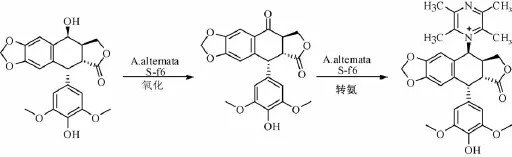
图5 具体反应过程(2.4)
3 鬼臼毒素C-4β位的结构修饰与作用
鬼臼毒素母核上4β位取代基结构的变化将显著影响其生物活性,且N-连接衍生物的活性明显高于O-和S-连接。目前,在保留C-4位取代的β构型、反式内酯环及C-4′位游离酚羟基的基础上,对鬼臼毒素C-4β位O原子转化成其生物等排体N原子后,再对该位置上的氨基或取代氨基进行修饰,正成为国内外的研究热点。
3.1 C-4β位酰胺类衍生物 这类衍生物通常是经酰胺键将鬼臼毒素与其它抗癌药物连接,修饰的原因主要有两个。一来,在肿瘤的临床治疗中,化疗药物剂量大,并且常需联合使用。二来,多效价、多靶标是目前设计药物广泛接受的手段,这类药物可以同时干预与疾病相关的多个靶标或者当细胞产生抗药性后改变靶点,能明显提高药物的选择性和活性。
Liu等[22]考虑到铂化合物对治疗睾丸癌等疾病的作用,并且卡铂和依托泊苷在临床上也常联合使用,于是设计了二氯卡铂共价结合到鬼臼毒素上,得到化合物7和8,它们均显示了对多种肿瘤细胞(如A-549、HeLab HCT、Hep-G2、K562、ADM/K562)的中等抑制作用。
Kamal等[4]发现,脲衍生物 (如二芳基取代脲)对肾细胞癌和不可切除的肝细胞癌具有抑制作用,同时也是潜在的DNA拓扑异构酶Ⅰ和Ⅱ的抑制剂。然后,对鬼臼毒素进行了结构改造,合成了4β-[4'-(1-(芳基)脲基)苯甲酰胺]鬼臼毒素类衍生物,其中化合物10~13对Colo-205细胞的活性明显高于依托泊苷。另外,他们还根据顺式芪对许多癌细胞的抑制活性,设计了3种衍生物,再将其与鬼臼毒素连接,结果发现,化合物14~16抑制
Guo等[24]利用2-[1-(4-氯苄基)-1H-吲哚-3-基]-2-氧代-N-(吡啶-4-基)乙酰(Indibulin)对微管蛋白的抑制作用及其在多药抗性方面能与鬼臼毒素的互补性,合成了一系列具有Indibulin环的4β-N-取代鬼臼毒素衍生物,其中化合物17能通过下调P糖蛋白的表达,从而对耐依托泊苷的KBV200 cell和K562/A02 cell具有较好抑制作用(化合物10~17的具体结构见图6)。
3.2 C-4β位不对称脲类衍生物 Li等[16]合成了20种4β-酰基硫脲取代的鬼臼毒素衍生物,而硫脲是在许多肿瘤发生的过程中起着关键作用的金属离子配体,还能作为色氨酸激酶的抑制剂。结果发现,化合物18~22活性略高于依托泊苷,对鼻咽癌耐药株KBVIN的活性也很明显,证明了C-4β位N取代鬼臼毒素衍生物具有克服多药抗性的潜在作用 (化合物18~22的具体结构见图7)。
3.3 C-4β位杂环类衍生物 Ren等[33]发现,噁二唑作为酰胺、酯、脲的生物电子等排体,常被用于修饰多种抗菌、抗肿瘤、抗炎、抗惊厥药物。他们通过6步反应制得4β-1,3,4-噁二唑-2-氨基-鬼臼毒素衍生物23~31,并测定了它们对DU-145、SGC-7901、A549、SH-SY5Y、HepG2、HeLa、L929、Vero的活性。结果发现,大多数化合物至少对一种测试肿瘤细胞产生了较好的抑制作用 (化合物23~31的具体结构见图8)。
3.4 C-4β位取代仲胺类衍生物 在药物中,一些具有特殊结构骨架结构的化合物显示出明显高于其它化合物的活性,这类结构被称为优势结构,将其适当改造后可呈现出不同的生物活性。Liu等[14]及Wang等[26]基于活性较好的化合物5和6在活性及多抗药性方面的优势,合成出了一
系列在N和杂环之间有1个C原子的化合物,其中化合物32~41对Hela、K562和K562/AO2均具有较好的抑制作用,IC50在10-6~10-8mol/L之间 (化合物32~41的具体结构见图9)。
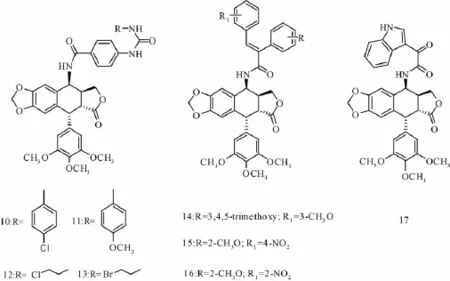
图6 化合物10~17的具体结构

图7 化合物18~22的具体结构

图8 化合物23~31的具体结构
3.5 C-4β位芳胺类衍生物 药物对一种拓扑异构酶的抑制可引起另一种的过度表达,最终使细胞产生耐药性。为避免上述现象,Ye[34]等合成了将拓扑异构酶Ⅰ型抑制剂喜树碱衍生物和拓扑异构酶Ⅱ抑制剂鬼臼毒素通过C-4β位N连接的化合物42-45。结果发现,虽然结合物活性降低,但达到对拓扑异构酶Ⅰ、Ⅱ的双重抑制的效果,可以为后期拓扑异构酶的双相抑制剂设计提供参考 (化合物42~45的具体结构见图10)。
4 展望
鬼臼毒素主要存在于小檗科足叶草属、鲜黄莲属、山荷叶属和八角莲属植物中[35],在自然界分布广泛,并具有多种药理活性,而且其4β-糖基取代衍生物依托泊苷和替尼泊苷是成熟的临床药物。因此,新型4β-鬼臼毒素衍生物,尤其是4β-N取代物的合成和药理活性分析是目前研究的热点,现已取得了很多有价值的成果。随着计算机辅助药物设计及药物构效关系的发展和深入,开发出更适用于临床的高活性4β-N取代鬼臼毒素衍生物将成为可能。
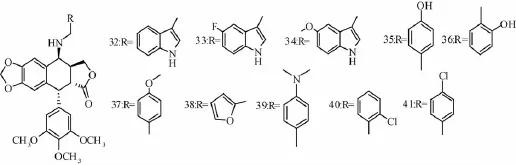
图9 化合物32~41的具体结构
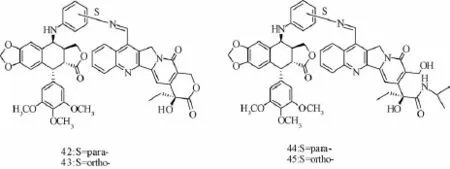
图10 化合物42~45的具体结构
[1] Imbert T F.Discovery of podophyllotoxins[J].Biochimie,1998,80(3):207-222.
[2] Kier L B,Fitzgerald D B,Burgett S.Isolation of podophyllotoxin from Callitrus drummondii[J].JPharm Sci,1963,52(5):502-503.
[3] Wang Y,Shao Y,Wang Y,et al.Synthesis and quantitative structure-activity relationship(QSAR)study ofnovel isoxazoline and oxime derivatives of podophyllotoxin as insecticidal agents[J].JAgric Food Chem,2012,60(34):8435-8443.
[4] Kamal A,Suresh P,Ramaiah M J,et al.4β-[4′-(1-(Aryl)ureido)benzamide]podophyllotoxins as DNA topoisomerase Iand IIαinhibitors and apoptosis inducing agents[J].Bioorg Med Chem,2013,21(17):5198-5208.
[5] Xu H,Wang Q,Guo Y.Stereoselective synthesis of 4α-alkyloxy-2-α/β-bromopodophyllotoxin derivatives as insecticidal agents[J].Chemistry,2011,17(30):8299-8303.
[6] Chen S W,Gao Y Y,Zhou N N,et al.Carbamates of 4′-demethyl-4-deoxypodophyllotoxin:synthesis,cytotoxicity and cell cycle effects[J].Bioorg Med Chem Lett,2011,21(24):7355-7358.
[7] Gordaliza M,García P A,del Corral JM,et al.Podophyllotoxin:distribution,sources,applications and new cytotoxic derivatives[J].Toxicon,2004,44(4):441-459.
[8] Carter S K,Slavik M.Investigational drugs under study by the United States National Cancer Institute[J].Cancer Treat Rev,1976,3(1):43-60.
[9] Fields S Z,Igwemezie L N,Kaul S,etal.Phase Istudy ofetoposide phosphate(etopophos)as a 30-minute infusion on days 1,3,and 5[J].Clin Cancer Res,1995,1(1):105-111.
[10] Baldwin E L,Osheroff N.Etoposide,topoisomeraseⅡand cancer[J].Curr Med Chem Anticancer Agents,2005,5(4):363-372.
[11] Jordan A,Hadfield JA,Lawrence N J,etal.Tubulin asa target for anticancer drugs:agents which interact with the mitotic spindle[J].Med Res Rev,1998,18(4):259-296.
[12] Buchardt O,Jensen R B,Hansen H F,et al.Thermal chemistry of podophyllotoxin in ethanoland a comparison of the cytostatic activity of the thermolysis products[J].JPharm Sci,1986,75(11):1076-1080.
[13] Pitts S L,Jablonksy M J,Duca M,et al.Contributions of the D-Ring to the activity of etoposide against human topoisomerase IIα:potential interactionswith DNA in the ternary enzyme-drug-DNA complex[J].Biochemistry,2011,50(22):5058-5066.
[14] Liu J,Cao B,Gao Y,et al.Design,synthesis,and antitumor activity of novel podophyllotoxin derivatives as potent anticancer agents[J].JAsian Nat Prod Res,2013,15(9):985-992.
[15] You Y.Podophyllotoxin derivatives:current synthetic approaches for new anticancer agents[J].Curr Pharm Des,2005,11(13):1695-1717.
[16] LiW Q,Wang X L,Qian K,et al.Design,synthesis and potent cytotoxic activity of novel podophyllotoxin derivatives[J]. Bioorg Med Chem,2013,21(8):2363-2369.
[17] 许明录,汤 波.DNA拓扑异构酶I和II的结构特性及抑制剂的研究进展[J].河南科技学院学报,2009,32(2):32-38.
[18] Hande K R.Etoposide:four decades of development of a topoi-
someraseⅡinhibitor[J].Eur JCancer,1998,34(10):1514-1521.
[19] Xiao L,ZhaoW,LiHM,etal.Design and synthesisof thenovel DNA topoisomeraseⅡinhibitors:Esterification and amination substituted 4′-demethylepipodophyllotoxin derivates exhibiting anti-tumor activity by activating ATM/ATR signaling pathways[J].Eur JMed Chem,2014,80:267-277.
[20] Pichichero M E,Avers C J.The evolution of cellularmovement in eukaryotes:the role ofmicrofilaments and microtubules[J]. Subcell Biochem,1973,2(1):97-105.
[21] 尚 海,潘 莉,杨 澍,等.微管蛋白抑制剂的研究进展[J].药学学报,2010,45(9):1078-1088.
[22] Liu X,Zhang L L,Xu X H,et al.Synthesis and anticancer activity of dichloroplatinum(Ⅱ)complexes of podophyllotoxin[J].Bioorg Med Chem Lett,2013,23(13):3780-3784.
[23] Liu JF,Sang C Y,Xu X H,et al.Synthesis and cytotoxic activity on human cancer cells of carbamate derivatives of4β-(1,2,3-triazol-1-yl)podophyllotoxin[J].Eur J Med Chem,2013,64:621-628.
[24] Guo Y E,Chen H,Zuo S,etal.Synthesisand antitumor activity of novel podophyllotoxin derivatives againstmultidrug-resistant cancer cells[J].J Asian Nat Prod Res,2011,13(5):417-424.
[25] Kamal A,Suresh P,Janaki Ramaiah M,et al.Synthesis and biological evaluation of 4β-acrylamidopodophyllotoxin congeners as DNA damaging agents[J].Bioorg Med Chem,2011,19(15):4589-4600.
[26] Wang C N,Wu Z H,Zhao Y,et al.Synthesis and cytotoxicity evaluation of novel podophyllotoxin derivatives[J].Arch Pharm,2011,344(11):735-740.
[27] Yu Y P,Chen S Y,Wang Y G,et al.A facile and efficient synthesis of 4β-aminopodophyllotoxins[J].Tetrahedron Lett, 1999,40(10):1967-1970.
[28] Zhou X M,Wang Z Q,Chang JY,et al.Antitumor agents. 120.New 4-substituted benzylamine and benzylether derivatives of4′-0-demethylepipodophyllotoxin as potent inhibitors ofhuman DNA topoisomeraseⅡ[J].J Med Chem,1991,34(12):3346-3350.
[29] Hansen H F,Jensen R B,Willumsen A M,et al.New compounds related to podophyllotoxin and congeners:synthesis,structure elucidation and biological testing[J].Acta Chem Scand,1993,47(12):1190-1200.
[30] Kamal A,Kumar B A,Suresh P,et al.An efficient one-pot synthesis of benzothiazolo-4β-anilino-podophyllotoxin congeners:DNA topoisomerase-Ⅱinhibition and anticancer activity[J]. Bioorg Med Chem Lett,2011,21(1):350-353.
[31] Chen S W,Wang Y H,Jin Y,et al.Synthesis and anti-HIV-1 activities of novel podophyllotoxin derivatives[J].Bioorg Med Chem Lett,2007,17(7):2091-2095.
[32] Tang Y J,Zhao W,Li H M.Novel tandem biotransformation process for the biosynthesis ofa novel compound,4-(2,3,5,6-tetramethylpyrazine-1)-4′-demethylepipodophyllotoxin[J]. Appl Environ Microbiol,2011,77(9):3023-3034.
[33] Ren J,Wu L,Xin W Q,etal.Synthesis and biologicalevaluation of novel4β-(1,3,4-oxadia—zole-2-amino)-podophyllotoxin derivatives[J].Bioorg Med Chem Lett,2012,22(14):4778-4782.
[34] Ye D,Shi Q,Leung CH,et al.Antitumor agents 294.Novel E-ring-modified camptothecin-4β-anilino-4′-0-demethyl-epipodophyllotoxin conjugates as DNA topoisomerase I inhibitors and cytotoxic agents[J].Bioorg Med Chem,2012,20(14):4489-4494.
[35] 张 杰,周春山,刘 韶,等.小八角莲中鬼臼毒素的提取工艺研究[J].中药材,2007,30(1):103-104.
R284.3
A
1001-1528(2015)12-2733-06
10.3969/j.issn.1001-1528.2015.12.034
2015-05-06
张志华 (1978—),女,博士,研究方向为有机合成及天然产物的生物转化。E-mail:bridge1026@163.com
