扶手椅型缺陷硫化钼纳米带电子性质的第一性原理计算
2015-12-07欧阳方平彭盛霖贾治安
邵 妍 欧阳方平,,* 彭盛霖 刘 琦 贾治安 邹 慧,*
(1中南大学物理与电子学院, 先进材料超微结构与超快过程研究所, 长沙 410083;2中南大学粉末冶金研究院, 粉末冶金国家重点实验室, 长沙 410083)
扶手椅型缺陷硫化钼纳米带电子性质的第一性原理计算
邵 妍1欧阳方平1,2,*彭盛霖2刘 琦1贾治安1邹 慧1,*
(1中南大学物理与电子学院, 先进材料超微结构与超快过程研究所, 长沙 410083;2中南大学粉末冶金研究院, 粉末冶金国家重点实验室, 长沙 410083)
基于密度泛函理论的第一性原理计算, 研究了含空位缺陷的扶手椅型二硫化钼纳米带的电子性质. 发现缺陷会导致纳米带结构稳定性降低, 单空位钼缺陷和三空位缺陷使得纳米带从半导体变成金属性, 而单空位硫缺陷和两种双空位缺陷仅减小纳米带的带隙; 电子态密度和能带的本征态表明缺陷纳米带费米能级附近的杂质态主要是缺陷态的贡献. 研究了四类半导体性质的纳米带带隙与宽度的关系, 对于完整的纳米带, 带隙随宽度以3为周期振荡变化; 而引入空位缺陷后, 纳米带的带隙振荡不再具有周期且振荡幅度变小. 同时发现, 当缺陷的浓度变小后, 缺陷仅使纳米带的带隙减小, 不会使其变为金属性. 这些结果有望打开其在新型纳电子器件中的应用潜能.
二硫化钼; 纳米带; 空位缺陷; 第一性原理; 电子结构
1 引 言
作为一种新颖的二维层状类石墨烯材料,1过渡金属硫化物MoS2因其结构独特、性质新颖,2–4引起了各个领域研究者的广泛关注. 实验上已经使用化学气相沉积法(CVD)获得了大面积的多晶单层实验和计算结果均表明单层MoS2是具有类石墨烯结构的直接带隙半导体, 可作为场效应管中很好的通道材料,3,7同时在光电子学和纳电子学领域表现出了巨大的应用潜力.8–14Radisavljevic等9成功构建了开关比达1 × 108的MoS2基晶体管, 且其载流子迁移率为200 cm2V–1s–1, 有望用于集成电路中. Jiang和Li15甚至利用MoS2等过渡金属硫化物设计了隧道场效应晶体管, 其能耗较低, 未来有望应用于计算机中使其大幅度节能.
随着实验技术的进步, 在实验上已经成功合成准一维MoS2纳米带(MNRs).16,17与石墨烯纳米带类似, 单层MoS2转变为一维纳米带后, 其电子性质、磁学性质和输运性质依赖于边缘态.18–20研究发现,扶手椅型MoS2纳米带(AMoS2NR)是无磁性的直隙半导体, 锯齿型MoS2纳米带(ZMoS2NR)表现为有磁性的金属性.20,21研究者提出了很多调控其能带的方法, 比如原子吸附、22,23边缘氢化、22缺陷23,24等等,其中缺陷无疑是种很重要的方法. Shidpour等25指出ZMoS2NR边缘的单空位S原子缺陷会使其电子性质发生改变. 同时Li等26发现在ZMoS2NR边缘构造V型缺陷, 会使纳米带由金属转变为p型半导体. 值得注意的是, Jiang等27最近构造了MoS2基的隧道场效应晶体管, 发现在通道区的金属空位缺陷会引入带隙态从而减小隧道过渡区的有效长度使得MoS2隧道场效应晶体管的隧穿电流增加10倍. 然而, 目前的理论计算主要集中于对锯齿型MoS2纳米带缺陷的研究上. 鉴于此, 为了完善对MoS2纳米带缺陷影响的研究, 调节其性能使MoS2能够更加完美地应用于器件设计中, 本文选择在扶手椅型MoS2纳米带上构造单空位缺陷、双空位缺陷以及三空位缺陷, 对不同空位缺陷的AMoS2NR的稳定性与电子结构进行模拟研究, 计算了缺陷纳米带电子结构随纳米带宽度的变化趋势, 并分析引起变化的原因.
2 计算模型与方法

图1 扶手椅型MoS2纳米带(AMoS2NRs)模型示意图Fig.1 Model of armchair MoS2nanoribbon (AMoS2NRs)
采用与纳米带轴向平行的Mo-S链数(NA)表示纳米带的宽度, 选取NA= 12的AMoS2NR作为理论计算的基础模型. 如图1(a)所示, d表示纳米带原胞的晶格矢量长度, 为是MoS2的晶格常数). 选取图1(a)中较小黑色矩形框内结构作为这部分计算的完整纳米带原胞, 在此原胞基础上, 在其边缘中心位置去掉相应的原子形成五种空位缺陷纳米带, 根据移除原子的数目与种类, 图1(b–f)分别命名为单空位Mo缺陷纳米带(VMo/AMoS2NR)、三空位MoS2缺陷纳米带单空位S缺陷纳米带(VS/AMoS2NR)、双空位2S缺陷纳米带(V2S/AMoS2NR)以及双空位MoS缺陷纳米带(VMoS/AMoS2NR), 其中缺陷原子用小圈标记.
本文采用基于密度泛函理论(DFT)的第一性原理计算软件包ATK(Atomistix ToolKit)完成计算. 结构优化采用Broyden-Fletcher-Goldfarb-Shanno (BFGS)方法, 收敛标准为原子受力小于0.5 eVnm-1,交换关联势为PBE泛函形式的广义梯度近似(GGA).所有的自洽计算均在周期性边界条件下执行, 优化结构和电子自洽计算中选取1 × 1 × 11的k点网格.电荷与势能积分实空间网格的截断能为150Ry(里德堡), 利用线性四面体积分方法计算电子态密度时选取1 × 1 × 41的k点网格. 与纳米带垂直的两个方向设置了略大于1 nm的真空层来避免相邻纳米带之间的相互影响.
3 计算结果与讨论
3.1 空位缺陷对扶手椅型纳米带电子结构的影响
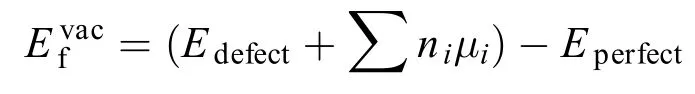
其中Edefect和Eperfect分别代表有缺陷的和完整纳米带的总能量, µi(i = S, Mo)代表单晶i原子的能量, ni代表缺陷纳米带中移除的i原子的数目. VMo/AMoS2NR、 VMoS/AMoS2NR、VS/AMoS2NR和 V2S/AMoS2NR的缺陷形成能分别是13.92、17.47、23.01、5.73、11.45 eV, 均为正值, 说明空位缺陷的形成是一个吸热过程, 因此会导致其结构的稳定性降低, 同时发现, 含Mo原子的三种缺陷比仅含S原子的两种缺陷的缺陷能大, 表明含Mo的缺陷更能破坏纳米带的稳定性.

图2 AMoS2NRs优化后的能带结构与几何结构图(俯视图)Fig.2 Band structures and geometries (top view) of AMoS2NRs after optimization
接着计算了不同缺陷纳米带的能带图及优化后的几何结构图, 如图2所示. 比较六种纳米带优化后的键长, 差异主要体现在缺陷周围的原子上, 其余键长大致相同. 在VMo/AMoS2NR中, 由于一个Mo原子的缺失, Mo―S共价键断裂, 使其周围的四个S原子存在孤对电子, 化学键重构, 导致缺陷周围四个Mo―S键键长变短, 如图2(b)所示; 同理, VS/AMoS2NR和VMoS/AMoS2NR缺陷周围的Mo―S键均变短. 在结构中, 缺失的三个原子所在的六元环结构严重扭曲变形, 两个S原子向纳米带内侧收缩, 且与邻近Mo原子的键长变小. 而在V2S/AMoS2NR中, 由于两个S原子的缺失导致两个Mo原子互成金属键. 从结构的成键状态来看, 仅去掉S原子形成的两种结构的稳定性较高, 与缺陷能得出的结论一致.
从能带图中知AMoS2NR的带隙宽度为0.421 eV, 与文献中相符.28我们的结果显示, 与完整纳米带对比, 五种缺陷纳米带在费米能级附近均存在类似杂质能级的能带, 其中VMo/AMoS2NR和AMoS2NR各自有两条穿过费米能级, 表现出金属特性; VS/AMoS2NR、V2S/AMoS2NR以及VMoS/ AMoS2NR均保持半导体性质, 不同的是VS/AMoS2NR和V2S/AMoS2NR的带隙大小接近AMoS2NR, 分别为0.416和0.410 eV, 而AMoS2NR的带隙(0.281 eV)小于AMoS2NR. /AMoS2NR, (d) VS/AMoS2NR, (e) V2S/AMoS2NR, (f) VMoS/AMoS2NR. The zero of the energy scale is set at the Fermi level.

图3 AMoS2NRs的电子态密度(DOS)和Mo, S原子的分轨道态密度图(PDOS)Fig.3 Densities of states (DOS) of AMoS2NRs and partial DOS (PDOS) of Mo, S atoms
为了进一步理解纳米带的电子结构, 比较了各个缺陷纳米带的Mo原子(在图2中标记为Mo-1、Mo-2)和S原子(在图2中标记为S-1、S-2)与AMoS2NR相对应原子的局域态密度(PDOS), 如图3所示. 其中Mo-1和Mo-2的差异是Mo-1位于离缺陷位置较远处而Mo-2是缺陷旁边的原子; 同理, S-1与S-2的差异也是如此. VMo/AMoS2NR和的局域态密度显示, Mo-1和S-1均表现出半导体行为, 与完整纳米带的Mo原子和S原子相似, 而Mo-2和S-2表现金属行为, 因此这两种缺陷结构的金属性主要来源于缺陷周围的原子. 同样计算了VS/AMoS2NR、V2S/AMoS2NR和VMoS/AMoS2NR的PDOS, 发现在此三类缺陷纳米带中Mo、S原子的PDOS的展宽和分裂与完整纳米带中相对应原子PDOS基本相似, 所有的原子均表现出半导体行为, 然而, PDOS的峰值的位置表现出了差异, 在图3(d)和3(e)中, 缺陷周围原子(S-2、Mo-2)与离缺陷位置较远的原子(S-1、Mo-1)的PDOS比较, 发现其峰值的位置几乎重叠,因此VS/AMoS2NR和V2S/AMoS2NR的带隙接近AMoS2NR; 而在图3(f)所示VMoS/AMoS2NR的PDOS中, 缺陷附近处的Mo-2与S-2的PDOS在距离费米能级最近处的两个波峰均向费米能级靠近, 因此导致VMoS/AMoS2NR与AMoS2NR相比较, 价带顶上移, 导带底下移, 导致其带隙小于AMoS2NR. PDOS计算结果说明空位缺陷对邻近缺陷的原子的影响较大, 缺陷态是影响了纳米带电子性质的主要因素.
为了证明缺陷态影响了纳米带的边缘态, 导致了杂质能级的引入, 图4计算了各个纳米带费米能级附近的几条特殊能带的本征态. 可以看到, 在AMoS2NR中, 最高价带(HVB)和最低导带(LCB)(即A1和A2)的电子本征态由相同的原子轨道组成, 这使得会在两个态之间产生一个非零的跃迁能量,18因此AMoS2NR的最高价带和最低导带不能形成简并的态, 并且随着它们靠近会出现能带相互排斥现象.然而在VMo/AMoS2NR和中, 分析了穿过费米能级的能带(分别标记为B1、B2和C1、C2)的本征态, 发现其原子轨道主要集中于缺陷周围, 因此可知这两种纳米带的金属性主要缘于缺陷态, 与PDOS得出的结论一致; 在VS/AMoS2NR、V2S/AMoS2NR和VMoS/AMoS2NR的能带图中, 可看出相对于AMoS2NR, 均在价带顶和导带底附近引入了杂质能带, 因此对这三种纳米带相关本征态的分析,除了价带顶(分别标记为D1、E1、F1)和导带低(分别标记为D2、E2、F2)之外, 还计算了Γ点离价带顶和导带底最近的两条杂质能带(杂质能带分别标记为D3、D4和E3、E4以及F3、F4)的本征态, 与AMoS2NR一致, 三条能带HVB和LCB的本征态由相同的原子轨道组成, 因此均表现出半导体性质, 而杂质能带主要是缺陷态引起的.(d) VS/AMoS2NR, (e) V2S/AMoS2NR, (f) VMoS/AMoS2NR. The zero of the energy scale is set at the Fermi level.

图4 纳米带特殊能带的本征态(eigenstates)Fig.4 Eigenstates of some special energy bands of nanoribbons
3.2 纳米带宽度对带隙的影响
计算了在不同宽度下纳米带的能带结构, 发现当纳米带宽度变化时, 每种缺陷对AMoS2NR的调控结果与前面一致, 即VMo和使纳米带变为金属性, 其他三种空位缺陷只能调控纳米带的带隙, 依旧表现出半导体性质.
因为量子限域效应和边缘效应的影响,28AMoS2NR的带隙随Mo―S链数的增加以3为周期振荡变化(如图5嵌入图所示), 这一规律与扶手椅型石墨烯纳米带相似,29由前面所知, VS、V2S和VMoS的引入只是调控AMoS2NR的带隙宽度, 但是, 是否这三种缺陷的引入会破坏这种周期性振荡? 为此, 计算了这三种半导体性质的缺陷纳米带的带隙随纳米带宽度的变化, 结果如图5所示. 对于VS/AMoS2NR、V2S/AMoS2NR和VMoS/AMoS2NR, 由于边缘态受到了缺陷态的影响, 振荡均变得杂乱无规律, 振荡周期不存在. 同时, 与前面得到的结论一致, 在相同的纳米带宽度下, VS/AMoS2NR和V2S/AMoS2NR的带隙大致相同, 且接近相应宽度AMoS2NR的带隙, 相对而言, VMoS/AMoS2NR的带隙较小, 与AMoS2NR的带隙差距较大.
3.3 缺陷浓度对纳米带能带结构的影响
纳米带的链数NA不变, 原胞宽度增加为2d(即选取图1(a)中较大矩形框内结构作为AMoS2NR原胞),在原胞的边缘中心位置构造五种缺陷, 此时纳米带的缺陷浓度相较于3.1节中的减小一半. 然后计算了这五种缺陷纳米带的能带图, 结果如图6所示.(c) VS/AMoS2NR, (d) V2S/AMoS2NR, (e) VMoS/AMoS2NR. The zero of the energy scale is set at the Fermi level.

图6 缺陷浓度变小后AMoS2NRs的能带图Fig.6 Energy band structures of AMoS2NRs after the defect concentrations decreased
从图6中可以看出, 当浓度减小, 所有的缺陷仅调控纳米带的带隙宽度而不打破其半导体性质. VMo/AMoS2NR和的带隙大小仅仅是0.084和0.069 eV, VMoS/AMoS2NR的带隙为0.279 eV, VS/AMoS2NR和V2S/AMoS2NR的带隙分别为0.406和0.381 eV. 可得到与前面一样的结论, 含Mo原子的缺陷对纳米带的影响较大, 其中VMo和对 AMoS2NR带隙的调控作用非常显著, 两种纳米带的带隙均极小, VMoS也使带隙减小了0.142 eV; 而仅含S原子的两种缺陷对带隙大小的调控较为弱小, 可推断, 当缺陷浓度变得更小时, 缺陷的引入只会调控完整纳米带的带隙大小, 含有金属Mo原子的缺陷对带隙大小的调控作用一定比仅有S原子的空位缺陷强. 同时计算了在此缺陷浓度下不同宽度的各个缺陷纳米带的带隙, 发现其带隙震荡不具有周期性,与前面的结果一致.
4 结 论
采用第一性原理计算方法, 研究了空位缺陷对扶手椅型硫化钼纳米带电子结构的影响. 通过在纳米带边缘移除原子模拟不同的缺陷结构, 计算几何结构与电子性质. 首先, 能带结果表明, 单空位Mo缺陷和三空位MoS2缺陷的引入使MoS2纳米带转变为金属性, 单空位S缺陷、双空位2S缺陷及双空位MoS缺陷会调控AMoS2NR的带隙大小, 说明空位缺陷的引入会改变纳米带的能带结构; 其局域态密度和能带本征态显示, 纳米带电子性质的改变是由缺陷态影响了边缘态导致的. 然后, 探讨了缺陷纳米带的电子性质与纳米带宽度的关系, 即使纳米带宽度不同, 同种缺陷对能带结构的调控是一致的, 但是由于缺陷态影响了纳米带, 与完整纳米带不同,半导体性质的三种缺陷纳米带的带隙不具有周期振荡性质. 最后, 计算缺陷浓度减小后纳米带的能带结构, 缺陷浓度变小后, 仅调控纳米带的带隙大小. 计算结果表明引入缺陷可以调控AMoS2NR的能带结构, 其中部分缺陷造成纳米带从半导体到金属性的转变有望使其应用与场效应晶体管中, 而缺陷对带隙大小的调节使其可能在一些半导体功能器件中得到应用. 研究结果可为将来过渡金属硫化物纳米带的研究及应用提供一定意义的理论指导.
(1)Wang, Q. H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J. N.; Strano, M. S. Nat. Nanotechnol. 2012, 7 (11), 699. doi: 10.1038/nnano.2012.193
(2)Kuc, A.; Zibouche, N.; Heine, T. Phys. Rev. B 2011, 83 (24), 245213. doi: 10.1103/Physrevb.83.245213
(3)Lebegue, S.; Eriksson, O. Phys. Rev. B 2009, 79 (11), 115409. doi: 10.1103/Physrev.79.115409
(4)Mak, K. F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T. F. Phys. Rev. Lett. 2010, 105 (13), 136805. doi: 10.1103/Physrevlett. 105.136805
(5)He, Q. Y.; Wu, S. X.; Gao, S.; Cao, X. H.; Yin, Z. Y.; Li, H.; Chen, P.; Zhang, H. ACS Nano 2011, 5 (6), 5038. doi: 10.1021/nn201118c
(6)Zeng, Z. Y.; Yin, Z. Y.; Huang, X.; Li, H.; He, Q. Y.; Lu, G.; Boey, F.; Zhang, H. Angew. Chem. Int. Edit. 2011, 50 (47), 11093. doi: 10.1002/anie.201106004
(7)Balendhran, S.; Ou, J. Z.; Bhaskaran, M.; Sriram, S.; Ippolito, S.; Vasic, Z.; Kats, E.; Bhargava, S.; Zhuiykov, S.; Kalantar-Zadeh, K. Nanoscale 2012, 4 (2), 461. doi: 10.1039/c1nr10803d
(8)Benameur, M. M.; Radisavljevic, B.; Heron, J. S.; Sahoo, S.; Berger, H.; Kis, A. Nanotechnology 2011, 22 (12), 125706. doi: 10.1088/0957-4484/22/12/125706
(9)Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Nat. Nanotechnol. 2011, 6 (3), 147. doi: 10.1038/nnano. 2010.279
(10)Feng, W. X.; Yao, Y. G.; Zhu, W. G.; Zhou, J. J.; Yao, W.; Xiao, D. Phys. Rev. B 2012, 86 (16), 165108. doi: 10.1103/Physrevb.86.165108
(11)Ma, Y. D.; Dai, Y.; Guo, M.; Niu, C. W.; Lu, J. B.; Huang, B. B. Phys. Chem. Chem. Phys. 2011, 13 (34), 15546. doi: 10.1039/c1cp21159e
(12)Butler, S. Z.; Hollen, S. M.; Cao, L. Y.; Cui, Y.; Gupta, J. A.; Gutierrez, H. R.; Heinz, T. F.; Hong, S. S.; Huang, J. X.; Ismach, A. F.; Johnston-Halperin, E.; Kuno, M.; Plashnitsa, V. V.; Robinson, R. D.; Ruoff, R. S.; Salahuddin, S.; Shan, J.; Shi, L.; Spencer, M. G.; Terrones, M.; Windl, W.; Goldberger, J. E. ACS Nano 2013, 7 (4), 2898. doi: 10.1021/nn400280c
(13)Novoselov, K. S.; Fal'ko, V. I.; Colombo, L.; Gellert, P. R.; Schwab, M. G.; Kim, K. Nature 2012, 490 (7419), 192. doi: 10.1038/nature11458
(14)Georgakilas, V.; Otyepka, M.; Bourlinos, A. B.; Chandra, V.; Kim, N.; Kemp, K. C.; Hobza, P.; Zboril, R.; Kim, K. S. Chem. Rev. 2012, 112 (11), 6156. doi: 10.1021/cr3000412
(15)Jiang, X. W.; Li, S. S. Appl. Phys. Lett. 2014, 104 (19), 193510. doi: 10.1063/1.4878515
(16)Li, Q.; Newberg, J. T.; Walter, E. C.; Hemminger, J. C.; Penner, R. M. Nano Lett. 2004, 4 (2), 277. doi: 10.1021/nl035011f
(17)Wang, Z. Y.; Li, H.; Liu, Z.; Shi, Z. J.; Lu, J.; Suenaga, K.; Joung, S. K.; Okazaki, T.; Gu, Z. N.; Zhou, J.; Gao, Z. X.; Li, G. P.; Sanvito, S.; Wang, E. G.; Iijima, S. J. Am. Chem. Soc. 2010, 132 (39), 13840. doi: 10.1021/ja1058026
(18)Georgiou, T.; Jalil, R.; Belle, B. D.; Britnell, L.; Gorbachev, R.V.; Morozov, S. V.; Kim, Y. J.; Gholinia, A.; Haigh, S. J.; Makarovsky, O.; Eaves, L.; Ponomarenko, L. A.; Geim, A. K.; Novoselov, K. S.; Mishchenko, A. Nat. Nanotechnol. 2013, 8 (2), 100. doi: 10.1038/Nnano.2012.224
(19)Kou, L. Z.; Tang, C.; Zhang, Y.; Heine, T.; Chen, C. F.; Frauenheim, T. J. Phys. Chem. Lett. 2012, 3 (20), 2934. doi: 10.1021/jz301339e
(20)Lukowski, M. A.; Daniel, A. S.; Meng, F.; Forticaux, A.; Li, L. S.; Jin, S. J. Am. Chem. Soc. 2013, 135 (28), 10274. doi: 10.1021/ja404523s
(21)Wei, J. W.; Ma, Z. W.; Zeng, H.; Wang, Z. Y.; Wei, Q.; Peng, P. AIP Adv. 2012, 2 (4), 042141. doi: 10.1063/1.4768261
(22)Cooper, R. C.; Lee, C.; Marianetti, C. A.; Wei, X. D.; Hone, J.; Kysar, J. W. Phys. Rev. B 2013, 87 (3), 035423. doi: 10.1103/Physrevb.87.035423
(23)Li, T. S. Phys. Rev. B 2012, 85 (23), 235407. doi: 10.1103/Physrevb.85.235407
(24)Li, J. W.; Medhekar, N. V.; Shenoy, V. B. J. Phys. Chem. C 2013, 117 (30), 15842. doi: 10.1021/jp403986v
(25)Shidpour, R.; Manteghian, M. Nanoscale 2010, 2 (8), 1429. doi: 10.1039/b9nr00368a
(26)Li, X. M.; Long, M. Q.; Cui, L. L.; Xiao, J.; Xu, H. Chin. Phys. B 2014, 23 (4), 047307. doi: 10.1088/1674-1056/23/4/047307
(27)Jiang, X. W.; Gong, J.; Xu, N.; Li, S. S.; Zhang, J. F.; Hao, Y.; Wang, L. W. Appl. Phys. Lett. 2014, 104 (2), 023512. doi: 10.1063/1.4862667
(28)Li, Y. F.; Zhou, Z.; Zhang, S. B.; Chen, Z. F. J. Am. Chem. Soc. 2008, 130 (49), 16739. doi: 10.1021/ja805545x
(29)Ouyang, F. P.; Xu, H.; Wei, C. Acta Phys. Sin. 2008, 57, 1073. [欧阳方平, 徐 慧, 魏 辰. 物理学报, 2008, 57, 1073.]
First-Principles Calculations of Electronic Properties of Defective Armchair MoS2Nanoribbons
SHAO Yan1OUYANG Fang-Ping1,2,*PENG Sheng-Lin2LIU Qi1JIA Zhi-An1ZOU Hui1,*
(1Institute of Super-microstructure and Ultrafast Process in Advanced Materials, School of Physics and Electronics, Central South University, Changsha 410083, P. R. China;2Powder Metallurgy Research Institute, State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, P. R. China)
We investigated the electronic properties of armchair MoS2nanoribbons with vacancy defects using a first-principles method based on density functional theory. It was found that defects reduced the stability of armchair MoS2nanoribbons. Mo vacancies and MoS2triple vacancies can both change the band structures of nanoribbons from semiconductor to metallic, whereas S vacancies, 2S divacancies, and MoS divacancies only decrease the bandgap. The densities of states and eigenstates of the nanoribbons indicated that impurity bands near the Fermi level basically contributed to the defect states. The relationships between the bandgap and width of four types of semiconducting nanoribbons were simulated. Nanoribbons with no defects have a bandgap that oscillates with width in a period of three, but the bandgap changes nonperiodically for nanoribbons with S vacancies, 2S divacancies, and MoS divacancies. We also found that when the concentration of defects decreased, the vacancy defects did not destroy the nanoribbonsemiconducting behavior but only decreased the bandgap. These results open up possibilities for MoS2nanoribbon applications in novel nanoelectronic devices.
Molybdenum disulfide; Nanoribbon; Vacancy defect; First-principles; Electronic structure
O641
10.3866/PKU.WHXB201510132
Received: May 7, 2015; Revised: October 8, 2015; Published on Web: October 13, 2015.
*Corresponding authors. ZOU Hui, Email: zouhui1115@163.com; Tel: +86-15173115723. OUYANG Fang-Ping, Email:
oyfp04@mails.tsinghua.edu.cn; Tel: +86-13973119546.
The project was supported by the National Natural Science Foundation of China (51272291, 21103232, 11104356), Natural Science Fund for
Distinguished Young Scholars of Hunan Province of China (2015JJ1020), State Key Laboratory of Powder Metallurgy, China (2014091907), and Central South University Research Fund for Faculty, China (2013JSJJ022).
国家自然科学基金(51272291, 21103232, 11104356), 湖南省杰出青年科学基金项目(2015JJ1020), 粉末冶金国家重点实验室科研课题重点项目(2014091907)和中南大学教师研究基金(2013JSJJ022)资助项目
©Editorial office of Acta Physico-Chimica Sinica
