基于分子对接和QSAR方法预测B-Raf II型抑制剂活性
2015-12-07刘海春张艳敏徐金星潇徐安阳陈亚东
刘海春 卢 帅 冉 挺 张艳敏 徐金星 熊 潇徐安阳 陆 涛,2,* 陈亚东,*
(1中国药科大学理学院, 南京 211198; 2中国药科大学, 天然药物活性组分与药效国家重点实验室, 南京 210009)
基于分子对接和QSAR方法预测B-Raf II型抑制剂活性
刘海春1卢 帅1冉 挺1张艳敏1徐金星1熊 潇1徐安阳1陆 涛1,2,*陈亚东1,*
(1中国药科大学理学院, 南京 211198;2中国药科大学, 天然药物活性组分与药效国家重点实验室, 南京 210009)
B-Raf激酶在促分裂素原活化蛋白激酶(MAPK)信号转导通路中起着重要作用, 已被确定为癌症治疗非常有吸引力的靶标. 新型高效B-Raf抑制剂的开发成为癌症治疗的一个热门研究领域. 本文以结构多样的B-Raf II型抑制剂为研究对象, 联合应用分子对接和定量构效关系(QSAR)模型研究其定量构效关系去探讨抑制活性的起源. 两个主题作为研究重点: 生物活性构象和描述符. 首先对分子对接方法(Glide、Gold、LigandFit、Cdocker和Libdock)进行准确性评价, 后将研究的对象分子对接到B-Raf活性位点并获得生物活性构象. 基于准确的对接结果, 计算得到16个打分评价函数和21个能量描述符, 以此构建定量构效关系模型. QSAR结果表明模型具有高度精确的拟合和强的预测能力(模型同时探讨了对抑制活性有重要影响的描述符, 结果表明打分评价函数(G_Score, -ECD, Dock_Score, PMF)与能量描述符(S(hb_ext), DE(int), Emodel)对抑制活性影响非常大. 通过虚拟筛选和QSAR模型理论预测, 一些新的具有潜在抑制活性的化合物作为B-Raf II型抑制剂被获得. 上述信息对于进一步设计新颖高效的B-Raf II型抑制剂提供了有用的指导.
B-Raf II型抑制剂; 分子对接; 打分评价函数; 对接能量描述符; 定量构效关系模型
1 引 言
促分裂素原活化蛋白激酶(MAPK)信号转导通路在细胞的生长、分化和增殖中起着重要作用.1Raf是MAPK信号转导通路中一个重要的丝氨酸-苏氨酸蛋白激酶家族, 其激活突变能够促进细胞的转化与增殖, 因此与肿瘤发生密切相关. Raf激酶家族有3 个亚型: A-Raf、B-Raf和C-Raf (Raf-1). 与ARaf和C-Raf相比, B-Raf有更高的激酶活性. 目前已确定B-Raf被激活后的体细胞突变率在黑色素瘤中高达50%–70%, 卵巢癌中达35%, 甲状腺癌中达30%, 结肠癌中达10%.2最常见的B-Raf突变发生在临近保守的DFG (Asp-Phe-Gly) 基序的600位残基,即谷氨酸取代了缬氨酸(V600E). B-RafV600E具有癌基因的特点, V600E突变的B-Raf的催化活性比野生型B-Raf (B-RafWT)提高了500倍.3–5
通过B-Raf激酶抑制剂与其蛋白结合的晶体复合物分析, 根据DFG序列及α-C螺旋位置的不同, BRaf激酶抑制剂可分为I型、II型和I1/2型.6目前已有4个B-Raf激酶抑制剂上市, 包括II型抑制剂Sorafenib和Regorafenib,7–10以及I1/2型抑制剂Vemurafenib和Dabrafenib11–16(图1). 据报道I型和I1/2型抑制剂可以异常激活B-RafWT或者MAPK通路中Raf-1, 引起肿瘤的二次增长. 而II型抑制剂因能够抑制多种激酶的活性, 包括Raf激酶、VEGFR-2、VEGFR-3、PDGF-β、C-KIT和Ret等, 可以逆转这种耐药性.17–19
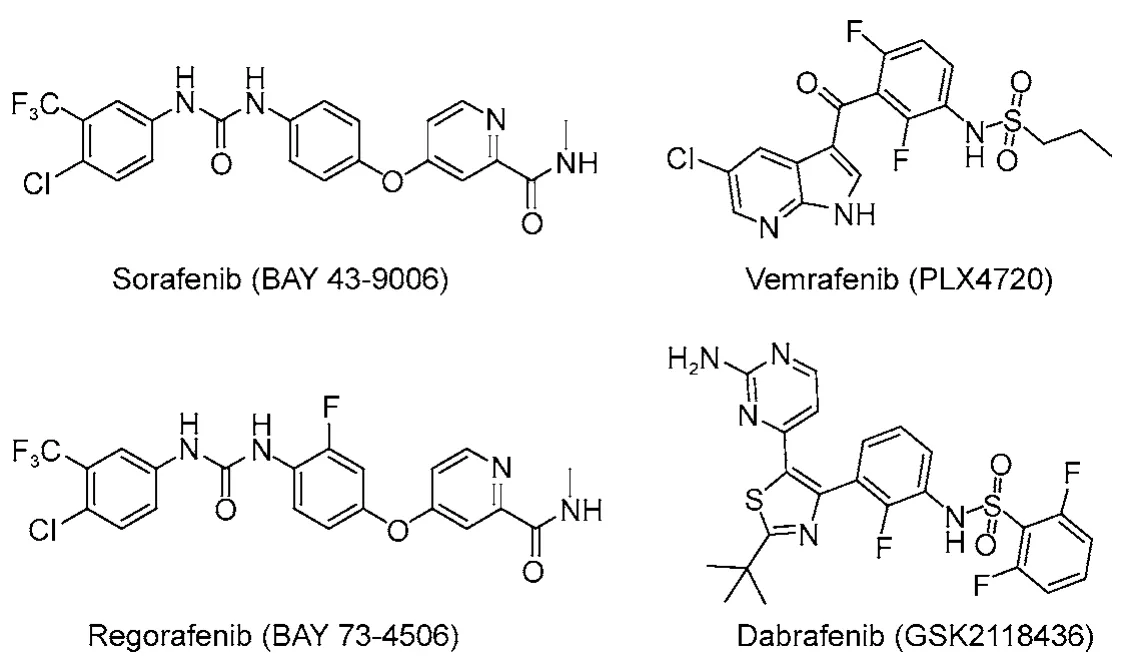
图1 已上市的B-Raf激酶抑制剂的结构Fig.1 Structures of the approved B-Raf kinase inhibitors
近年计算机辅助药物分子设计(CADD)在新药开发中得到了越来越多的应用, 定量构效关系(QSAR)和对接(DOCK)是CADD的两个主要工具.分子对接方法是将小分子化合物放入大分子蛋白靶标活性口袋中, 预测小分子与靶标的结合模式,同时应用打分函数评估其与大分子的结合自由能.分子对接目前已相对成熟, 但绝非完美, 仍有大量的问题有待进一步研究. 目前打分函数只能粗略评估结合自由能及预测生物活性.20–23幸运的是, QSAR方法具有准确可靠评估结合自由能, 预测新的先导物生物活性的能力. QSAR通过理论计算已知结构的先导物的物理化学参数进行统计分析, 找出结构与其生物效应(如生物利用度、活性或毒性等)的定量关系, 建立结构活性模型并以此指导药物分子设计. QSAR方法应用日益广泛, 具有较高的预测准确度, 但也遭遇到了不少问题. 目前选择和设计合适的描述符参数是QSAR研究的一大热点. 随着计算能力的提高, 计算化学也将提供更为准确的参数, 为进行准确的QSAR研究奠定基础.24,25
为准确预测分子结合自由能提高虚拟筛选打分排序, 本研究结合分子对接和QSAR方法的技术力量建立以对接产生的打分函数和能量描述子为自变量的QSAR模型. 先是分子对接准确性评价, 对Glide、Gold、Cdocker、LigandFit和Libdock26–28对接策略进行可靠性验证. 在准确的对接基础上, 对结构多样的B-Raf II型抑制剂进行对接建立正确的结合模式, 产生分子对接评价参数, 包括16个打分评价函数和21个对接产生的能量描述符. 利用一元线性及多重线性(MLR)回归方法对打分评价函数及能量描述符和实验活性之间的相关性进行统计分析, 建立QSAR模型.29,30MLR-QSAR模型获得了预测活性和实验pIC50值之间的高相关性系数并具有良好的预测能力. 应用具有良好预测能力的MLRQSAR模型得到多个具有较好的预测活性且骨架新颖的化合物可作为潜在的B-Raf先导化合物. 分子对接联合QSAR模拟方法为合理设计和优化新的具有潜在活性的B-Raf II型抑制剂提供了新的策略.
2 实验材料和方法
2.1 B-Raf晶体复合物结构准备
从RCSB蛋白质结构数据库(PDB)选择最具代表性也是最早发表的B-Raf晶体复合物(PDB ID: 1UWH, 分辨率为0.295 nm, 配体为Sorafenib)作为对接模板.3复合物结构用Discovery Studio (DS)28进行预处理, A链及其配体被选定保留为对接目的. 将Sorafenib从复合物中提取出来, 除去所有水分子, 使用Sybyl31将配体和受体分别加氢加电荷进行结构能量最小化.
2.2 训练集和测试集的准备
B-Raf抑制剂数据来源于Binding Database (http://www.bindingdb.org/bind/index.jsp), 收集有BRaf抑制活性的化合物, 删除重复的I型和I1/2型结构及IC50≥ 10 μmolL–1的化合物, 最终选择70个具有明显活性变化梯度(IC50最小2.5 nmolL–1, 最大8800nmolL–1)和结构多样性的II型化合物用于模型构建和评价. 通常数据集随机被分为训练集和测试集.本文为提高QSAR模型可靠性和预测能力, 采用化学指纹图谱相似度聚类分析方法合理划分数据集.32,33化学指纹图谱聚类分析是一种将数据集划分为小区域的集群定义划分方法. 通过化学指纹图谱最大不同, 一些有代表性的分子被选为簇的中心,剩余分子被分配到最近的聚类中心找到相应的集群. 通过DS计算化学指纹图谱, Tanimoto系数为描述符描述分子相似度, 同时考虑活性梯度选择, 数据集被分为训练集38分子和测试集32分子(表中带星号化合物). 图2显示划分的训练集分子和测试集分子(蓝色小球)在化学空间的投影, 进一步说明数据集划分的合理性. 表1显示所有化合物结构和IC50值.
2.3 分子对接、打分函数和描述符
分子对接程序Glide、Gold、LigandFit、Cdocker和Libdock被应用, 16个打分函数和21个对接描述符被选择进行QSAR研究. 16个打分函数包括LigandFit的十一个打分函数(Dock_Score、PMF、PLP1、PLP2、Jain、Ligscore-1、Ligscore-2、PMF04、Ludi1、Ludi2、Ludi3), Gold的二个打分函数(Glod_Score和Chem_Score), 以及Glide的G_Score、Cdcoker的-Cdocker Interaction Energy和Libdock的Libdockscore; 对接描述符分别来源于不同的对接打分功能, 十一个来源于G_Score, 四个来源于Gold_Score, 六个来源于Chem_Score (表2).
2.3.1 Glide对接
Glide是近似为完全系统搜索配体的构象、取向和空间位置的对接软件. Glide对接过程中, 蛋白质的结构是刚性的, 而小分子的构象是柔性的. 蛋白准备模块Protein Preparation Wizard在OPLS-2005分子力场下准备受体; 以晶体复合物中的配体位置为对接中心, Receptor Grid Generation生成格点文件; Ligpre准备配体及其抑制剂. Ligand Docking进行分子对接, 选择标准精度(SP)柔性对接, 每个配体保留10个构象, 其它参数保持默认值. 选取打分最优的构象用于进一步研究, G_Score和十一个对接描述符被获得.
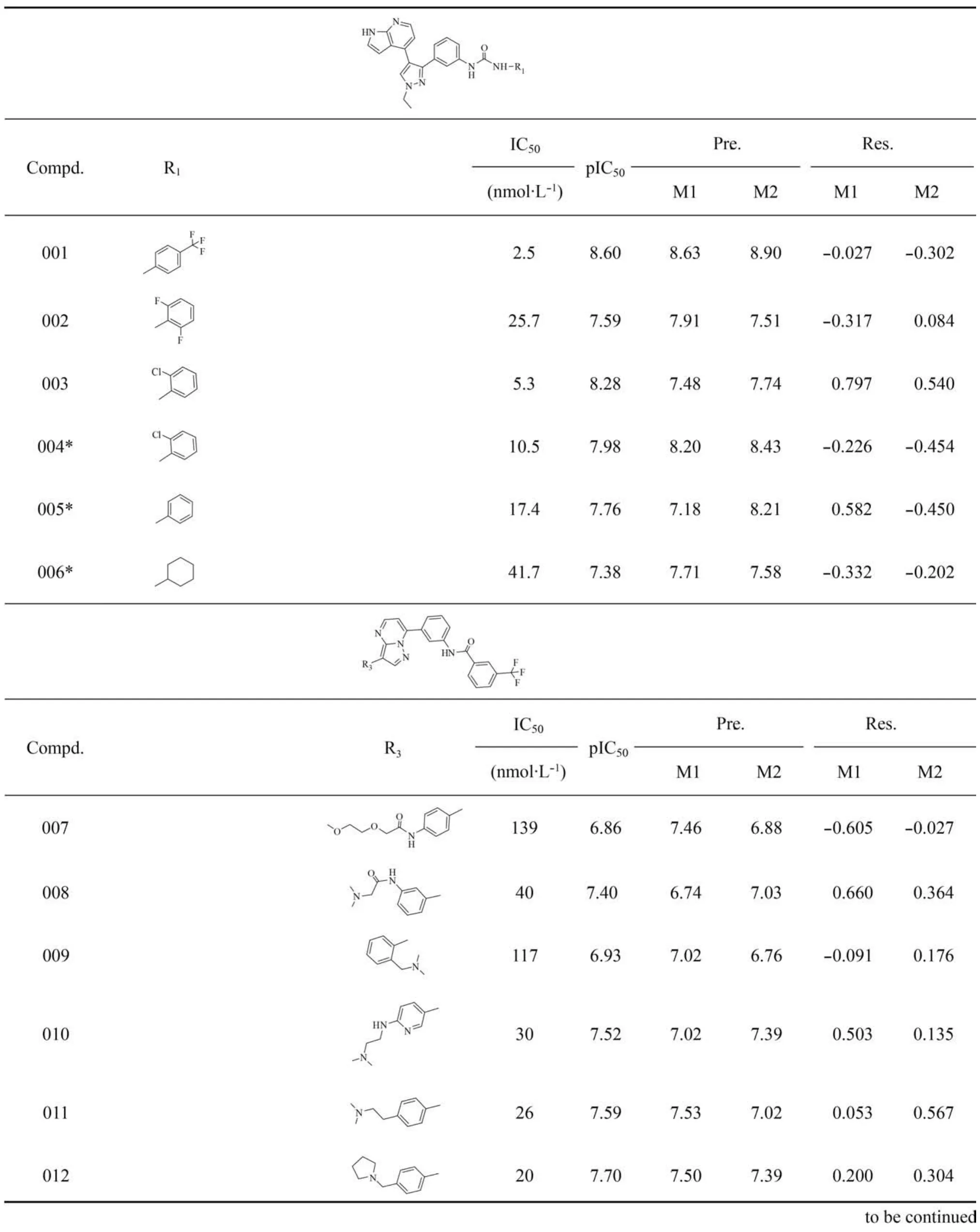
表1 B-Raf抑制剂的训练集和测试集分子结构, 实验活性值与QSAR模型(模型M1及M2)预测活性值(Pre.)及残差(Res.)Table1 Molecular structures, actual activities vs predicted activities (Pre.) and residuals (Res.) of QSAR models (M1 and M2) for the training set and the test set of B-Raf inhibitors





*compounds in the test set
2.3.2 Gold对接
Gold (Genetic Optimisat ion for Ligand Docking)程序是应用遗传算法在局部柔性蛋白中寻找柔性配体构象的自动柔性对接程序. 可以充分搜索构象空间, 找到相互作用的最优模式. 本文采用Gold_ Score和Chem_Score打分函数得到分子相互作用的拟合度得分(Fitness score, 函数值大小用来判断分子对接情况的好坏, 函数值越大则分子间相互作用越强, 形成的包合物能量越低, 越稳定)数值, 同时计算得到多种对接能量描述符. 结合位点用共结晶配体坐标位置来定义. 各参数保持缺省值, 每个配体保留10个构象.
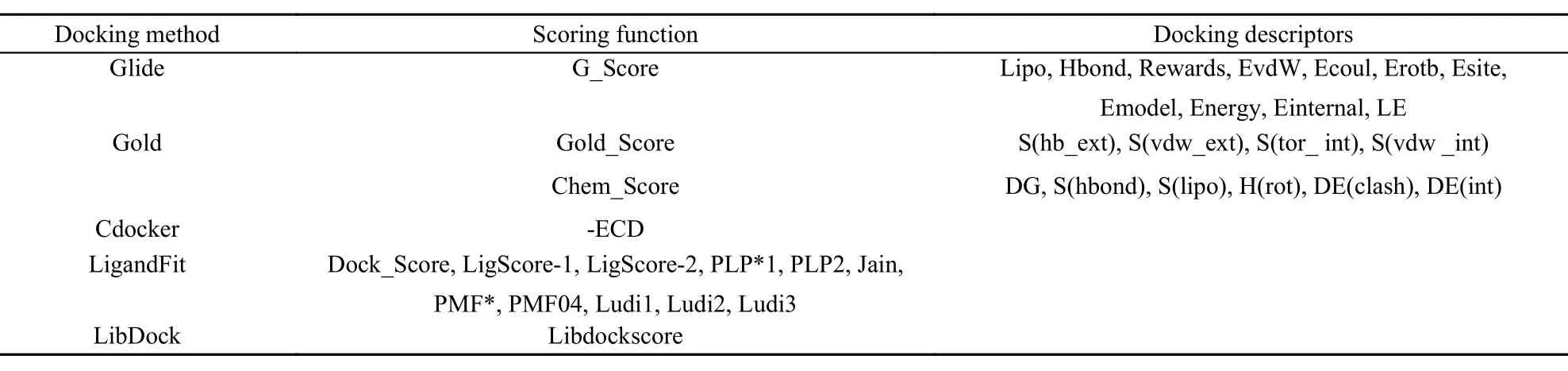
表2 分子对接方法, 打分评分函数及生成的各种描述符Table2 Molecular docking methods, scoring functions, and the various descriptors used for generating models
2.3.3 Cdocker对接
Cdocker是基于CHARMm力场的柔性对接程序, 设定蛋白质构象保持不变, 配体构象发生改变,计算配体分子和受体之间的非键相互作用, 以-Cdocker Interaction Energy(-ECD)表示受体和配体间结合自由能. 分子对接选取PDB晶体结构1UWH为受体模型, 通过加氢及CHARMm力场对其进行优化. 以配体原子来定义结合位点, 其范围半径都采用自动生成值, 将配体分子置入结合位点球中, 变更配体构象, 进行多构象分子对接. 将这些不同构象的配体分子与蛋白质视为一个整体复合物,采用CHARMm力场和退火模拟算法分别优化这些复合物的能量. 每个化合物对接后都得到10个构象,综合考虑其能量打分值(-ECD)及结合状态选取最优构象进行分析.
2.3.4 LigandFit 对接
LigandFit对接具备了受体分子活性位点的自动寻找和确认、构象柔性的多配体对接以及基于力场的相互作用打分评价的功能. LigandFit设定活性位点内关键的氨基酸上的基团相互作用作为筛选标准, 要求被对接的小分子发生相应的基团匹配,形成氢键, 疏水相互作用等等. 研究基于B-Raf晶体复合物中的配体Sorafenib定义对接结合位点, 应用蒙特卡洛算法对配体的构象空间进行取样, 以基于力场参数的方法进行受体和配体分子相互作用的评价. 使用Ligscore对虚拟筛选结果进一步评价, 包括LigScore-1、LigScore-2、PLP1、PLP2、Jain、PMF等在内的多种不同算法的打分函数.
2.3.5 LibDock 对接
LibDock是基于特征的对接(Site Feature Docking), 在将小分子放入到结合腔内之前需对结合腔内部的特征(热点区域“hot spot”, 即小分子配体要满足的化学特征)进行计算. LibDock预先生成化合物的低能量构象, 对接过程中不再生成新的分子构象. LibDock有自己独特的评分系统, 也可以使用PLP进行评分. 本研究以B-Raf晶体蛋白1UWH为受体, ATP结合位点为对接中心, 半径为1.5 nm, 每次保留30个构象, 配体构象产生方法为best, Hotspots为100, 其他参数默认, 对接结果以Libdockscore排序.
2.4 线性回归模型
本研究以不同的对接打分函数及对接产生的能量描述子作为独立变量, 利用一元线性和多元线性回归(MLR)算法构建强大的QSAR模型. 综合考虑对接打分函数和化合物与激酶的结合状态选取其最优构象. 对化合物生物活性值与其最优构象对接打分函数及能量描述子先进行一元线性回归, 分别得到一元线性回归方程. 同时运用TSAR软件34对打分函数及描述符进行MLR分析, 选出影响化合物抗癌活性的重要独立变量, 建立MLR模型. 一些统计指标, 如复相关性系数(r2)、交互验证相关系数检验值(F)、均方根误差(S)、测试集复相关系数评估模型的质量, 模型的拟合能力检验包括复相关系数和均方根误差, 模型的内部预测能力验证采用留一交叉验证法的CV), 模型的外部预测能力验证通过系数来评估.35,36

表3 Top3对接构象均方根偏差(RMSD)值验证对接可靠性Table3 Root mean square deviation (RMSD) values of Top3 configurations obtained through docking for docking reliability validated
3 结果与讨论
3.1 分子对接研究
3.1.1 对接可靠性验证
本工作的主要目的是利用不同对接程序产生的评分函数及描述符产生基于结构的QSAR模型,以对确定新的有效的B-Raf抑制剂起指导作用. 生成模型之前, 验证分子对接方法的可靠性是非常重要和必要的. 采用B-Raf及其配体Sorafenib复合物的X射线结构1UWH执行此验证. 通过Glide、Gold、LigandFit、Cdocker和Libdock将配体Sorafenib还原对接到激酶结合口袋, 验证对接方法能否再现结合底物在晶体结构中的构象. 对接后的分子构象与晶体结构中配体分子构象的均方根偏差(RMSD)作为评价标准, 排名前Top3最佳对接(相应最高打分或最低能量)构象进行验证考虑, 一般RMSD不超过0.20nm即认为对接还原成功. 可接受的对接结果(表3显示RMSD在0.0292–0.2154 nm)显示对接构象在结合位点内具有相似的结合位置和方向, 与晶体构象类似(图3显示Top1对接结果), 表明上述对接方法能成功在B-Raf晶体结构中还原再现结合底物的构象.
3.1.2 B-Raf II型抑制剂对接结果
70个具有生物活性B-Raf II型抑制剂分子(表1)被选择进行分子对接和特殊的QSAR模型研究.对所有化合物进行几何优化, 用上述对接方法对接到B-Raf (PDB 1UWH)晶体结构模板的结合位点. 基于对接打分评价函数及同B-Raf激酶之间的相互作用确定化合物的最佳对接构象. II型B-Raf激酶抑制剂作用模式表现为: (1)与DFG基序天冬氨酸(Asp)和α-C螺旋谷氨酸(Glu)形成一对关键性的氢键; (2)与铰链区半胱氨酸(Cys)上的NH和羰基形成1–2个氢键; (3)与变构位点附近的疏水口袋形成疏水作用; (4)铰链区附近有一个溶剂可及区(见图4).根据对接结果研究, 所有的化合物都能够充分准确对接, 基本满足上述作用模式, 整体取向一致, 展示对接模型良好的质量.

图3 Sorafenib共晶体构象(绿色)与Top1最佳对接构象(其他颜色)在B-Raf活性部位叠加图Fig.3 Superimposition of co-crystallized Sorafenib (green) and redocked Top1 conformation by multiple docking (the other colors) in the active site of B-Raf
3.2 相关模型的计算和验证
以Glide、Gold、LigandFit、Cdocker和Libdock对接方法计算得到的打分评价函数和能量描述子(表2)作为QSAR建模的自变量函数, 抑制剂分子的–lgIC50(pIC50)作为因变量函数用来构建对接打分函数/能量描述符和B-Raf II型抑制剂pIC50的相关性模型.
3.2.1 一元线性回归模型
首先研究实验活性与单一对接打分评价函数及其能量描述子的定量构效关系, 建立一元线性回归模型, 计算其相关性系数r2. 结果列于表4, r2在0.002到0.622之间. 对于单一打分评价函数, G_Score给出最好的相关系数(r2= 0.622), Libdockscore给最差的相关系数(r2= 0.027). -ECD、Dock_Score、LigScore-1和LigScore-2等也具有相对较好的相关性系数, r2在0.50左右. 这一结果表明, QSAR模型基于柔性对接的打分函数比刚性对接打分函数更能获得高的相关系数r2. 因为通过柔性对接方法可以得到更准确的结合模式, 而基于正确的结合模式基础上的活性预测被认为是合理的. 进一步说明对接方法和蛋白质模板的验证是必要的. 对于单一能量描述符, Emodel给出最好的相关系数(r2= 0.471), H(rot)给最差的相关系数(r2= 0.002). Rewards和ENERGY的r2分别为0.261和0.272, Lipo, Hbond, LE, S(vdw_int)的r2也高于0.20. 说明对接产生的能量描述符也可能对估计准确的活性值是有用的. 一元线性回归模型显示每一个独立变量同实验活性之间具有一定的相关性. 然而其相关性较低,大部分相关系数r2小于0.50 (表4). 因此, 单一使用对接打分函数/能量描述符对于确定有效的抑制剂和预测准确的抑制活性具有明显的局限性.

图4 B-Raf抑制剂作用模式示意图Fig.4 Schematic diagram of interaction patterns of B-Raf kinase inhibitor
3.2.2 多重线性回归模型
一元线性回归模型的结果促使我们开发新的预测模型, 因此采用TSAR软件, 通过MLR方法对训练集分子对接得到的16个打分函数及21个对接描述符分别进行特定的QSAR研究, 得到相关的回归方程M1和M2. 对回归方程进行了测试和验证, 以评估模型的质量. 最终得到的多元线性回归模型与一元线性回归模型相比, 活性相关性及预测能力大大提高. M1和M2的相应统计参数如表5所示.
QSAR模型M1使用所有16个打分评价函数生成, 回归方程:
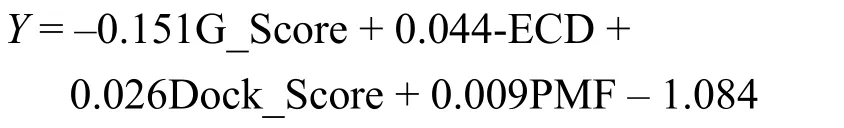
在QSAR建模中, 训练集样本直接参与模型的建立, 模型的拟合能力由相关系数(r2)、标准偏差(SD或S)来评价. r2主要体现模型对训练集样本数据的拟合能力, 通常认为r2越大越接近1说明模型的相关性越好, 拟合效果越好, QSAR模型质量越高. S表示数据与平均数据之间差值的平均数. S的大小表明数据的分散性, 同样表示模型的拟合能力, 越接近于零说明模型拟合效果越好. 一个模型对内部训练集样本有较好的拟合能力并不代表模型具有较好的预测能力. QSAR模型最主要目的是对未知化合物进行活性预测, 基于这个意义层面考虑, 模型的预测能力显得比拟合能力要重要. QSAR模型的稳健性也是必要的, 否则模型受到异常值的影响会出现抖动或者模型自身会存在机会相关问题. 为检验所建立QSAR模型的稳健性和内部预测能力, 采用“留一法”(LOO) 计算交叉验证(CV)系数评价模型稳健性和预测能力. 当大于0.5时, 通常认为所建立的模型具有较好的预测能力, 越接近于1说明稳定性越好, 预测能力越强.

表4 实验pIC50值与评分函数及描述符一元线性回归模型相关性系数Table4 Correlations of individual scoring function and descriptors with the experimental pIC50values

表5 QSAR模型M1和M2的相应统计参数Table5 QSAR modes M1, M2 and the corresponding statistics
综合考虑模型的拟合能力、稳健性及预测能力三个要素, Alexander Tropsha曾提出, 一个较好的QSAR模型需要满足下面两个条件: r2> 0.600,所得打分函数QSAR模型M1复相关系数r2、交互验证相关系数分别为0.852和 0.790, 说明模型具有良好的拟合、稳健性及内部预测能力, 此B-Raf II型抑制剂QSAR模型M1是可靠的.
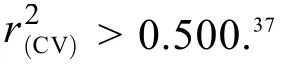

21个对接产生的表示配体和受体之间的静电力、范德华力、氢键以内部能量相互作用的描述符也被视为独立的变量生成QSAR模型M2, 回归方程:

模型对训练集和测试集中化合物的活性分别进行预测, 活性实验值和预测值以及残差见表1, 线性关系如图5和图6所示. 表1显示模型M1和M2训练集化合物的预测值与实验值绝大部分相差都很小,说明该模型有较强的拟合能力. 图5(a)和图6(a)所示为训练集实验值和预测值线性回归关系图, 由图可见实验值与预测值有较好的相关性, 进一步显示模型具有较好的拟合及内部预测能力. 同时表1显示模型M1和M2测试集化合物预测值与实验值相差也都很小, 说明该模型有较强的外部预测能力. 图5(b)和图6(b)所示为测试集的实验值和预测值线性回归关系图, 得到同训练集一样的结果, 说明模型具有好的外部预测能力可用来准确预测新抑制剂的生物活性.

图5 基于打分函数的QSAR模型M1实验活性值与预测活性值关系图Fig.5 Actual activity vs predicted activity using QSAR model M1 based on scoring functions
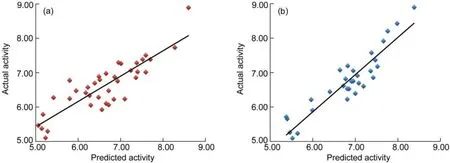
图6 基于打分函数的QSAR模型M2实验活性值与预测活性值关系图Fig.6 Actual activity vs predicted activity using QSAR model M2 based on docking descriptors
3.3 模型参数解释
模型M1包括4个独立的自变量, X1是G_Score, X2是-ECD, X4是Dock_Score, X8是PMF. G_Score基于经验的打分函数, 结合配体和受体之间的库仑和范德华相互作用能, 重点考虑氢键相互作用, 同时引入溶剂化作用. G_Score负的回归系数表明对亲和力有不利的贡献(G_Score打分为负值, 越小越有利). -ECD描述配体与受体的相互作用能, 包括范德华能与静电能量. Dock_Score是基于力场的打分函数, 包括配体的内能以及配体与受体的相互作用能,主要考虑静电和立体互补性对于结合能的贡献.PMF是基于知识的打分函数, 总结成对相互作用包括所有在受体-配体原子间相互作用连同金属离子相互作用和卤素的电极电位, 将其转换成距离依赖性Helmholtz自由能. -ECD、Dock_Score和PMF正的回归系数表明其与亲和力正相关, 对亲和力有积极有利的贡献. 模型M2中, 三个对接能量描述符S(hb_ext)、DE(int)、Emodel对活性预测有重要意义. S(hb_ext)为蛋白质-配体的氢键相互作用能, 有助于提高回归系数与其正相关, 对亲和力有有利的影响. DE(int)为内部扭转应变张力. Emodel是Glide对接得到的能量描述符,是对G_Score的拓展. Emodel结合了G_Score, 非键相互作用能和内部应变能. DE(int)和Emodel描述符与回归系数负相关,对亲和力有负贡献, 数值增大导致亲和力降低. QSAR模型M1和M2表明在QSAR研究中选择正确有效的描述符对建立良好模型的重要性. 同时结果表明通过改变某些重要描述符的大小可增强抑制活性.
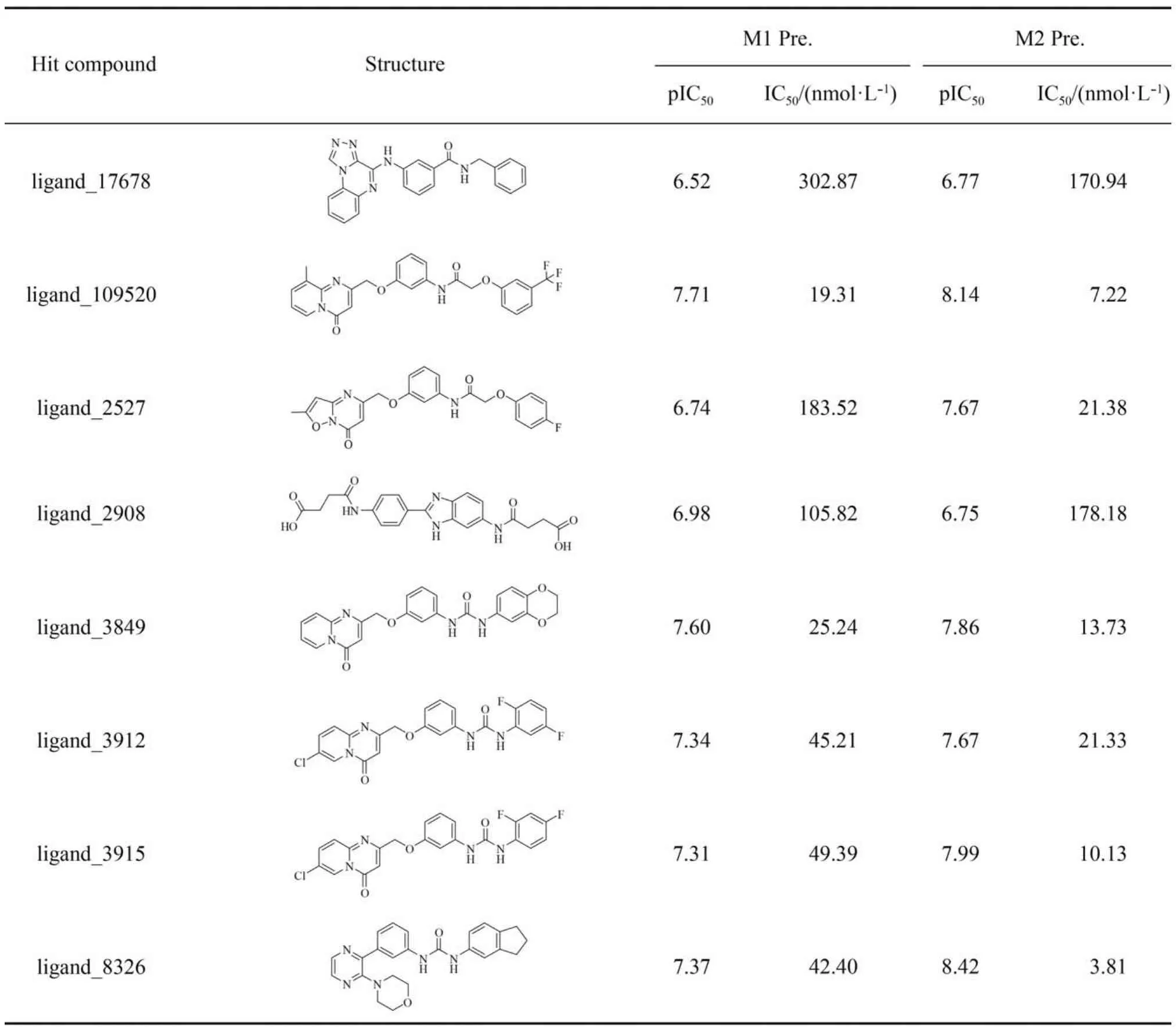
表6 苗头化合物化学结构及活性预测值Table6 Chemical structures and predicted activity values of the hit compounds
3.4 新化合物的筛选与活性预测
基于MLR模型, 采用分子对接对ChemDiv数据库进行虚拟筛选, 用QSAR模型进行活性预测. 应用Lipinski五规则进行初始过滤以减少数据库的样本数量. 首先使用一元线性回归得到最高相关系数的Glide进行对接虚拟筛选, 打分评价函数G_Score大于10.0且符合II抑制剂结合模式的化合物被选择进行深入研究. 对这些化合物进一步进行Gold, Cdocker、LigandFit、LibDock对接, 以得到其他打分评价函数和能量描述子进行QSAR模型活性预测.应用MLR-QSAR模型预测进行活性预测, 最终8个具有较好的预测pIC50值(pIC50> 6.5)且骨架新颖的化合物被选择作为潜在的先导化合物. 8个命中的苗头化合物化学结构和预测pIC50值列于表6, 仍有待进一步的实验确认和评价.
4 结 论
研究以定量构效关系和分子对接方法来预测B-Raf II型抑制剂的活性. 通过对接还原配体结晶构象验证分子对接方法(Glide、Gold、LigandFit、Cdocker和Libdock)的准确性. 结果表明, 对接方法可以预测正确的结合模式, 对接研究对象分子到BRaf的活性位点同样可以获得生物活性构象. 基于正确的结合模式, 计算得到对接打分函数及其能量相关的描述符. 利用一元线性及其多重线性回归方法对打分函数及能量描述符和实验活性之间的相关性进行统计分析, 找出结构与活性的定量关系, 建立QSAR模型. 一元线性模型显示描述符与实验活性的相关性较低. MLR-QSAR模型获得了高相关性系数及良好的内外部预测能力, 同时探讨了对抑制活性有重要影响的描述符. 应用MLR-QSAR模型的8个具有较好的预测活性且骨架新颖的化合物被选择作为潜在的先导化合物, 对于进一步设计新颖高效的B-Raf II型抑制剂提供了有用的指导. 分子对接联合QSAR模拟方法为合理设计和优化新的具有潜在活性的B-Raf II型抑制剂提供了新的策略. 该策略是一种很有前途的方法, 可用于新靶标的分子设计.
(1)Peyssonnaux, C.; Eychene, A. Biol. Cell 2001, 93, 53. doi: 10.1016/S0248-4900(01)01125-X
(2)Davies, H.; Bignell, G. R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M. J.; Bottomley, W.; Davis, N.; Dicks, E.; Ewing, R.; Floyd, Y.; Gray, K.; Hall, S.; Hawes, R.; Hughes, J.; Kosmidou, V.; Menzies, A.; Mould, C.; Parker, A.; Stevens, C.; Watt, S.; Hooper, S.; Wilson, R.; Jayatilake, H.; Gusterson, B. A.; Cooper, C.; Shipley, J.; Hargrave, D.; Pritchard-Jones, K.; Maitland, N.; Chenevix-Trench, G.; Riggins, G. J.; Bigner, D. D.; Palmieri, G.; Cossu, A.; Flanagan, A.; Nicholson, A.; Ho, J. W. C.; Leung, S. Y.; Yuen, S. T.; Weber, B. L.; Seigler, H. F.; Darrow, T. L.; Paterson, H.; Marais, R.; Marshall, C. J.; Wooster, R.; Stratton, M. R.; Futreal, P. A. Nature 2002, 417, 949. doi: 10.1038/nature00766
(3)Wan, P. T.; Garnett, M. J.; Roe, S. M.; Lee, S.; Niculescu-Duvaz, D.; Good, V. M.; Jones, C. M.; Marshall, C. J.; Springer, C. J.; Barford, D.; Marais, R. Cell 2004, 116, 855. doi: 10.1016/S0092-8674(04)00215-6
(4)Samowitz, W. S.; Sweeney, C.; Herrick, J.; Albertsen, H.; Levin, T. R.; Murtaugh, M. A.; Wolff, R. K.; Slattery, M. L. Cancer Res. 2005, 65, 6063. doi: 10.1158/0008-5472.CAN-05-0404
(5)Riesco-Eizaguirre, G.; Gutiérrez-Martínez, P.; García-Cabezas, M. A.; Nistal, M.; Santisteban, P. Endocr-Relat. Cancer 2006, 13, 257. doi: 10.1677/erc.1.01119
(6)Wang, X.; Kim, J. J. Med. Chem. 2012, 55 (17), 7332. doi: 10.1021/jm300613w
(7)Wilhelm, S. M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; Cao, Y.; Shujath, J.; Gawlak, S.; Eveleigh, D.; Rowley, B.; Liu, L.; Adnane, L.; Lynch, M.; Auclair, D.; Taylor, I.; Gedrich, R.; Voznesensky, A.; Riedl, B.; Post, L. E.; Bollag, G.; Trail, P. A. Cancer Res. 2004, 64, 7099. doi: 10.1158/0008-5472.CAN-04-1443
(8)Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R. A.; Schwartz, B.; Simantov, R.; Kelley, S. Nat. Rev. Drug Discov. 2006, 5 (10), 835. doi: 10.1038/nrd2130
(9)Wilhelm, S. M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C. A.; Schütz, G.; Thierauch, K. H.; Zopf, D. Int. J. Cancer 2011, 129 (1), 245. doi: 10.1002/ijc.v129.1
(10)Strumberg, D.; Schultheis, B. Expert Opin. Investig. Drugs 2012, 21 (6), 879. doi: 10.1517/13543784.2012.684752
(11)Chapman, P. B.; Hauschild, A.; Robert, C.; Haanen, J. B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; Hogg, D.; Lorigan, P.; Lebbe, C.; Jouary, T.; Schadendorf, D.; Ribas, A.; O'Day, S. J.; Sosman, J. A.; Kirkwood, J. M.; Eggermont, A. M. M.; Dreno, B.; Nolop, K.; Li, J.; Nelson, B.; Hou, J.; Lee, R. J.; Flaherty, K. T.; McArthur, G. A. N. Engl. J. Med. 2011, 364, 2507. doi: 10.1056/NEJMoa1103782
(12)Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Nature. Rev. Drug. Discov. 2012, 11, 873. doi: 10.1038/nrd3847
(13)Rheault, T. R.; Stellwagen, J. C.; Adjabeng, G. M.; Hornberger, K. R.; Petro, K. G.; Waterson, A. G.; Dickerson, S. H.; Mook, R. A., Jr.; Laquerre, S. G.; King, A. J.; Rossanese, O. W.; Arnone, M. R.; Smitheman, K. N.; Kane-Carson, L. S.; Han, C.; Moorthy, G. S.; Moss, K. G.; Uehling, D. E. ACS Med. Chem. Lett. 2013, 4, 358. doi: 10.1021/ml4000063
(14)Kim, D. H.; Sim, T. Arch. Pharm. Res. 2012, 35, 605. doi: 10.1007/s12272-012-0403-5
(15)Holderfield, M.; Deuker, M. M.; McCormick, F.; McMahon, M. Nat. Rev. Cancer 2014, 14, 455. doi: 10.1038/nrc3760
(16)Ackerman, A.; Klein, O.; McDermott, D. F.; Wang, W.; Ibrahim,N.; Lawrence, D. P.; Gunturi, A.; Flaherty, K. T.; Hodi, F. S.; Kefford, R.; Menzies, A. M.; Atkins, M. B.; Long, G. V.; Sullivan, R. J. Cancer 2014, 120, 1695. doi: 10.1002/cncr.28620
(17)Hartsough, E.; Shao, Y.; Aplin, A. E. J. Invest. Dermatol. 2014, 134, 319. doi: 10.1038/jid.2013.358
(18)Bucheit A. D.; Davies M. A. Biochem. Pharmacol. 2014, 87 (3), 381. doi: 10.1016/j.bcp.2013.11.013
(19)Zhang, F.; Lu, T.; Tang, W. F. Prog. Pharm. Sci. 2014, 38, 31. [张 帆, 陆 涛, 唐伟方. 药学进展, 2014, 38, 31.]
(20)Enyedy, I. J.; Egan, W. J. J. Comput. Aided Mol. Des. 2008, 22, 161. doi: 10.1007/s10822-007-9165-4
(21)Kroemer, R. T. Curr. Protein Pept. Sci. 2007, 8, 312. doi: 10.2174/138920307781369382
(22)Lin, J.; Li, Z. G.; Zou, J. W.; Lu, S. Y. Acta Chim. Sin. 2012, 11, 1309. [林 军, 李祖光, 邹建卫, 陆绍永. 化学学报, 2012, 11, 1309.]
(23)Li, X. D.; Hou, T. J.; Xu, X. J. Acta Phys. -Chim. Sin. 2005, 21, 504. [李旭东, 侯廷军, 徐筱杰. 物理化学学报, 2005, 21, 504.] doi: 10.3866/PKU.WHXB20050509
(24)Li, M. Y.; Xia, L. Journal of China Pharmaceutical University 2003, 34 (6), 586. [李敏勇, 夏 霖. 中国药科大学学报, 2003, 34 (6), 586.]
(25)Patel, H. M.; Noolvi, M. N.; Sharma, P.; Jaiswal, V.; Bansal, S.; Lohan, S.; Kumar, S. S.; Abbot, V.; Dhiman, S.; Bhardwaj, V. Med. Chem. Res. 2014, 23, 4991. doi: 10.1007/s00044-014-1072-3
(26)Maestro, Version 9.0; Schrödinger, L. L. C.: New York, 2011
(27)GOLD, Version 4.0; Astex Technology: Cambridge, 2001
(28)Discovery Studio Client, Version 2.5; Accelrys Inc.: San Diego, 2008
(29)Chen, L.; Chen, X. J. Mol. Graph. Model. 2012, 33, 35. doi: 10.1016/j.jmgm.2011.11.003
(30)Frączek, T.; Siwek, A.; Paneth, P. J. Chem. Inf. Model. 2013, 53, 3326. doi: 10.1021/ci400427a
(31)SYBYL, Version 7.1; Tripos Inc.: St. Louis, Missouri, 63144, U. S. A.
(32)Hassan, M.; Bielawski, J.; Hempel, J.; Waldman, M. Mol. Divers. 1996, 2 (1–2), 64. doi: 10.1007/BF01718702
(33)Matter, H.; Potter, T. J. Chem. Inf. Comput. Sci. 1999, 39 (6), 1211. doi: 10.1021/ci980185h
(34)TSAR Software, Version 3. 3; Accelrys Inc.: Oxford, England
(35)Roy, K. Expert Opin. Drug Discov. 2007, 2, 1567. doi: 10.1517/edc.2007.2.issue-12
(36)Qin, L. T.; Liu, S. S.; Xiao, Q. F.; Wu, Q. S. Environ. Chem. 2013, 32, 1205. [覃礼堂, 刘树深, 肖乾芬, 吴庆生. 环境化学, 2013, 32, 1205.]
(37)Tropsha, A.; Gramatica, P.; Gombar, V. K. QSAR Comb. Sci. 2003, 22, 69.
Accurate Activity Predictions of B-Raf Type II Inhibitors via Molecular Docking and QSAR Methods
LIU Hai-Chun1LU Shuai1RAN Ting1ZHANG Yan-Min1XU Jin-Xing1XIONG Xiao1XU An-Yang1LU Tao1,2,*CHEN Ya-Dong1,*
(1School of Basic Science, China Pharmaceutical University, Nanjing 211198, P. R. China;2State Key Laboratory of Natural Medcines, China Pharmaceutical University, Nanjing 210009, P. R. China)
B-Raf kinase plays an important role in the mitogen-activated protein kinase (MAPK) signaling transmission pathway and has been identified as an attractive target for cancer therapy. The exploitation of novel and efficient B-Raf inhibitors has become a hot research topic. In this study, we investigated quantitative structure–activity relationship (QSAR) to probe the origins of the inhibitory activities of B-Raf Type II inhibitors. We used structurally diverse B-Raf Type II inhibitors and an integrated docking and QSAR extended method. We focused mainly on two themes: bioactive conformations and descriptors. First, various molecular docking methods (Glide, Gold, LigandFit, Cdocker, and Libdock) were evaluated, and then all molecules were docked into the B-Raf active site to obtain the bioactive conformations. Secondly, based on the docking results, 16 scoring functions and 21 docking-generated energy-based descriptors were calculated to construct regressionmodels. The results gave highly accurate fitting and had strong predictive abilitiesThe important descriptors were also explored to elucidate the main factors influencing the inhibition activities. The models suggested that the scoring functions (G_Score, -ECD, Dock_Score, and PMF) and docking-generated energy-based descriptors (S(hb_ext), DE(int), and Emodel) were significant. Some new compounds that are potential B-Raf inhibitors were obtained through virtual screening and theoretical predictions using the established models. Such information is useful in guiding the design of novel and robust B-Raf Type II inhibitors.
B-Raf Type II inhibitor; Molecular docking; Scoring function; Docking-generated energybased descriptor; Quantitative structure–activity relationship model
O641
10.3866/PKU.WHXB201510134
Received: July 9, 2015; Revised: October 12, 2015; Published on Web: October 13, 2015.
*Corresponding authors. LU Tao, Email: lutao@cpu.edu.cn; Tel: +86-25-86185179. CHEN Ya-Dong, Email: ydchen@cpu.edu.cn; Tel: +86-25-86185163.
The project was supported by the National Natural Science Foundation of China (21102181, 81302634).
国家自然科学基金(21102181, 81302634)资助项目
©Editorial office of Acta Physico-Chimica Sinica
