邻二氯苯激发态动力学
2015-12-05李晓营王艳秋刘本康
李晓营 王 利 王艳秋 宋 哲 刘本康,*
(1辽宁师范大学物理与电子学院, 辽宁 大连 116029; 2大连民族大学物理与材料工程学院, 辽宁 大连 116600;3中国科学院大连化学物理研究所, 分子反应动力学国家重点实验室, 辽宁 大连 116023)
邻二氯苯激发态动力学
李晓营1王 利2,*王艳秋3宋 哲1刘本康3,*
(1辽宁师范大学物理与电子学院, 辽宁 大连 116029;2大连民族大学物理与材料工程学院, 辽宁 大连 116600;3中国科学院大连化学物理研究所, 分子反应动力学国家重点实验室, 辽宁 大连 116023)
利用飞秒分辨的激光泵浦-探测技术结合飞行时间质谱和光电子速度成像方法研究了邻二氯苯第一电子单重激发态(S1)的超快动力学. 邻二氯苯的S1态振动基态寿命为(651 ± 10) ps, 对应于S1振动基态向三重态的系间窜越过程. 邻二氯苯S1的高振动激发9a218a2对应两个衰减通道, 其中寿命为(458 ± 12) fs的超快过程对应于由处于振动激发的S1向高振动激发的基态(S0)发生的内转换过程, 而寿命为(90 ± 10) ps过程则对应由S1态向三重态(T1)的系间窜越过程, 电离产生的光电子能谱中长寿命的谱峰可能与系间窜越过程有关. S1态高振动态的旋轨耦合程度比低振动态的更强, 导致系间窜越过程更快.
邻二氯苯; 振动激发; 光电子成像; 光电子能谱
1 引 言
多原子分子激发态在光物理和光化学领域起着非常重要的作用, 相关动力学过程研究对深入理解相关化学反应和控制其发生过程有着重要意义.1处于激发态的多原子分子可以通过辐射方式和非辐射方式回到低能态, 后者如非绝热效应引起的内转换(IC)和系间窜越(ISC)等.2其中氯代芳烃因其广泛存在及其潜在的毒性风险, 特别是多卤代芳烃,历来被认为是剧毒环境污染分子二噁英的主要来源之一.3对于芳烃卤代化合物, 特别是多卤代芳烃的深入研究有着十分重要的意义, 一直是研究的热点体系.4–37氯苯的第一电子激发态及其阳离子基态的相关轨道信息及动力学信息已经有了相对较多的研究报道.4–8氯苯再次被氯原子取代后, 其电离能、分子构型等均发生较大变化, 对其激发态及其离子态光谱已经有很细致的研究.4–14
早在1942年Sponer18就研究了二氯苯的近紫外吸收谱, 对193和266 nm波长处光解动力学研究发现这类化合物在低激发能量下由单重激发态, 经内转换到基态热激发或经系间窜越到三重态, 从而使激发能定域在一个C-Cl 键上而解离.19–21EI-Sayed及其合作者22,23首先利用双波长皮秒激光研究了间位和对位二氯苯分子的电离解离过程并估计了激发态的寿命. 飞秒超快时间分辨使泵浦探测实验能够更接近激发态寿命的量级, Diau Eric等24以飞秒激光实时探测了邻、对、间二溴苯的C-Br 键断裂所需时间及产生的苯炔中间态的寿命. Kadi等25指出取代卤素原子质量越大, 因旋轨耦合导致的系间窜越速率越快, 激发态寿命越短. Yoshida等26用皮秒激光(160 ps)研究了氯苯和间二氯苯(m-DCB)激发态, m-DCB由于两个氯原子取代加强了旋轨耦合作用, 由于激光脉宽过大, 无法直接获得第一电子激发态的寿命; Deguchi等27实验估算出邻二氯苯分子(o-DCB)的S1态寿命应小于120 ps. 我们前期开展了对二氯苯(p-DCB)28和m-DCB29激发态动力学实验研究,在270 nm波长激发下, p-DCB和m-DCB的激发态显示出相似的动力学行为, 其激发态均表现出明显的量子拍频现象, 周期相近约为37 ps, 激发态总体寿命分别为(122 ± 4) ps和(109 ± 1) ps.28,29
和传统的光电子能谱技术相比, 飞秒时间分辨的光电子成像技术在提供高分辨的能谱信息的同时还可以提供相应的角度分布信息和时间分辨信息, 是研究激发态非绝热动力学的重要手段.30基于该实验方法, 国内外已有大量的关于复杂体系的超快动力学方面的优秀工作被陆续报道.31–35在此, 我们利用飞秒时间分辨的质谱和光电子成像技术, 研究了o-DCB第一单重电子激发态的振动基态和高振动态动力学过程, 明确阐释高振动态和振动基态动力学行为之间存在的显著差异.
2 实验部分
实验所采用的装置已经在文献36中报道过, 这里仅作简要说明. 自行研制的固态飞秒激光器的基频光输出脉冲宽度为70 fs, 中心波长814 nm, 光谱宽度约33 nm, 重复频率为20 Hz, 平均功率为150 mW. 基频光经过一个30 : 70分束片分为两束, 其中强度为30%的一束光经过延时平台后作为检测光(probe). 而另一束强度为70%的光进入光参量放大器(Quantronix/Light Conversion, TOPAS, Lithuania),光参量放大器的输出光经过倍频后获得波长为276和247 nm 的激发光(pump), 由于光参量放大过程的展宽, 光谱宽度约1.6 nm. 两束光经双色镜后同轴, 由透镜(焦距为35 cm)聚焦在分子束上. 两束光的偏振方向均垂直于分子束的传播方向, 且平行于离子成像检测器.
将0.4 MPa氦气通过盛有o-DCB (99%, Aldrich,没有进一步纯化)的样品池, 携带样品饱和蒸汽的混合气体经脉冲阀(Parker, General Valve series 9, USA, 喷口直径0.5 mm)喷射, 经过skimmer形成超音速分子束进入飞行时间质谱的电离区, 在离子速度成像透镜区域与飞秒激光相互作用并且被电离.37,38电离产生的离子或者电子经速度聚焦后经过40 cm长由μ合金屏蔽的无场飞行区后被离子成像检测器检测. 其中由离子成像检测器的二级微通道板(MCP, z-stack)上经过提取电路直接获得相应的飞行时间质谱, 由数字示波器(Tektronix Inc., TDS3054B)纪录和累加平均; 由CCD相机(LAVISION Inc., Imager QE, Germany)捕获荧光屏上的电子成像.
实验仪器的时间分辨是通过Xe原子非共振多光子电离泵浦-探测实验获得, 通过高斯拟合和退卷积处理得到本实验的仪器相关函数约为82 fs. 每张电子成像均为10000次激光累加的结果. 实验中获得的每个泵浦-探测时间延时的数据都是重复扫描20次的平均结果. 实验中仔细调节泵浦光和探测光的强度, 使得质谱中碎片离子信号几乎可以忽略不计, 以免碎片电离给光电子成像和能谱分析带来不确定因素.
3 结果与讨论
3.1 S1(1A1)态振动基态的超快动力学
o-DCB的第一电子激发态S1(1A1)振动基态为36231.9 cm–1(波长为275.9 nm, 4.492 eV), 绝热电离势的精确值为(73237 ± 6) cm–1((9.0799 ± 0.0006) eV).4,5波长为276 nm (带宽2 nm)光可以将o-DCB激发到S1(1A1)的振动基态. 检测光波长为814 nm, 单光子能量为1.529 eV. 处于S1(1A1)态振动基态的o-DCB需要吸收4个814 nm光子方可以被电离, 对应的总能量为10.58 eV. 图1是处于S1(1A1)态振动基态被814 nm电离产生的o-DCB母体离子信号随泵浦-探测延时时间变化情况, 其中实线为用相关函数和单指数衰减模型拟合和卷积的结果. o-DCB母体离子的单指数衰减寿命为(651 ± 10) ps, 对应于o-DCB的S1(1A1)态振动基态的非辐射跃迁过程. 该寿命值小于Shimoda等14的计算值1.4 ns, 大于Qin等39获得的S1(1A1)态高振动激发态(267 nm激发)的寿命(482 ps). 和文献39对比, 该结果也说明了随着第一电子激发态振动激发能量的增加, 衰减速率越发变快.我们实验获得的S1(1A1)态振动基态的非辐射跃迁过程比早期的理论预期要快.
和我们以前的p-DCB和m-DCB在270 nm飞秒激发态研究结果28,29不同的是, 当用275.9 nm飞秒激发时, o-DCB母体离子没有出现明显的量子拍频现象, 如图1所示. 在前期工作中,28,29飞秒激发光270nm由飞秒基频光通过三倍频技术产生, 在优化条件下飞秒脉宽展宽较小, 270 nm (37037 cm–1)处光谱半高全宽约为 5 nm, 可以同时将o-DCB激发至不同的振动频率和振动模式上. 根据光谱研究结果,4o-DCB主要的振动能级密集区域位于振动基态之上的1000 cm–1区域, 该区域包含了丰富的振动模式激发, 如11、18a1、18b1、7a16a1、6a19b1等, 宽带的270 nm可以实现对叠加态的激发. 而在本实验中, 飞秒激发光275.9 nm是通过光参量放大器产生的, 光参量放大器会伴随有一定的色散现象, 导致脉宽展宽及光谱带宽变窄, 相应半高全宽约为1.6 nm (约219 cm–1), 激发o-DCB第一电子激发态的振动基态时, 无法激发到附近的振动模式(36362 cm–1), 因此无法观测到清晰的由振动相干态或者振转耦合能级组成的相干重叠态随时间的演化过程, 即量子拍频现象.这可能是o-DCB第一电子激发态的振动基态寿命明显长于前期p-DCB和m-DCB激发态寿命的原因之一.
图2(a)是不同延迟时间的光电子图像. 利用pBasexl算法40,41对上述图像进行处理, 图2(a)左侧对应的是原始图像, 而右侧则是变换后的图像; 图2(b)中为相应的归一化光电子能谱. 从能谱结构上, 四个主要的谱峰分别标为A、B、C和D, 对应的能量分别为0.72、1.04、1.18和2.27 eV. 图2(b)显示在母体离子衰减过程中光电子能谱在结构上并未有明显的变化, 整体强度随时间变弱.

图1 S1(1A1)振动基态激发时o-DCB母体离子信号强度随延迟时间的变化Fig.1 Delay-time dependence of transient profile of o-DCB parent ion, excited at the vibrational ground state of the first excited electronic state S1(1A1)
o-DCB 被激发到S1(1A1)态振动基态后, 至少需要吸收4个814 nm光子, 体系总能量(hvpump+ 4hvprobe= 10.58 eV, 其中h为普朗克常数, vpump与vprobe分别代表激发光和探测光的频率)才可以高于第一电离势(9.08 eV). 图3为根据文献4,7,9,10,42,43总结出的o-DCB电子激发态、离子激发态和相关振动态的能级示意图. 电离产生的光电子能量为EK= hvpump+ 4hvprobe–Eion, 其中EK与Eion分别为光电子动能和离子能量. 对应的离子基态、离子第一激发态和离子第二激发态分别为X2B1、A2A2和B2B2, 对应各自的振动基态能量分别为9.08、9.53和10.70 eV. 图2(b)中峰A、B和C分别对应电离时离子态能量分别为9.86、9.54和9.40 eV. 如图3所示, 峰A、B和C对应已知的离子能级分别为9.77、9.53和9.40 eV, 对应的离子态依次为A2A2(v7+ v8)、A2A2(00)和X2B1(72101).43
图2(b)中峰D在图3中没有对应的能级, 峰D与A的能量差为1.55 eV, 与814 nm单光子能量(1.53 eV)接近, 因此峰D应当是5光子电离产生的, 对应的离子态与峰A相同.
3.2 S1(1A1)态高振动激发态的超快动力学
波长247 nm光可以将o-DCB激发至S1(1A1)态9a218a2振动模式,10用波长814 nm光检测, 获得o-DCB母体离子随延迟时间的衰减曲线, 如图4所示.图中实线是采用双指数衰减模型和响应函数卷积的最佳拟合结果; 插图为延时7.5 ps以内的快速变化过程及相应的拟合情形. 拟合结果显示S1(1A1)态 9a218a2振动态衰减过程可以分为两个过程, 一个是寿命为(458 ± 12) fs的超快过程, 另外一个是寿命为(90 ± 10) ps的慢过程, 分别对应于由S1向S0振动热带发生的内转换过程和S1–T1之间的系间窜越过程. 由该振动激发态发生的系间窜越显著快于由S1振动基态的系间窜越((651 ± 10) ps), 主要原因是高振动激发时S1与T1的振动耦合增加所致.14,15,21在o-DCB的S1(1A1)态振动基态激发过程中, 没有观察到相应的由S1向S0振动热带发生的快速内转换过程, 可能是由于在S1(1A1)态振动基态激发过程中没有足够的振动模式被激发, 从而导致与基态的高振动激发态之间的耦合效应较弱导致, 前人研究结果也标明这种耦合过程随激发能增加而迅速加强.15,21
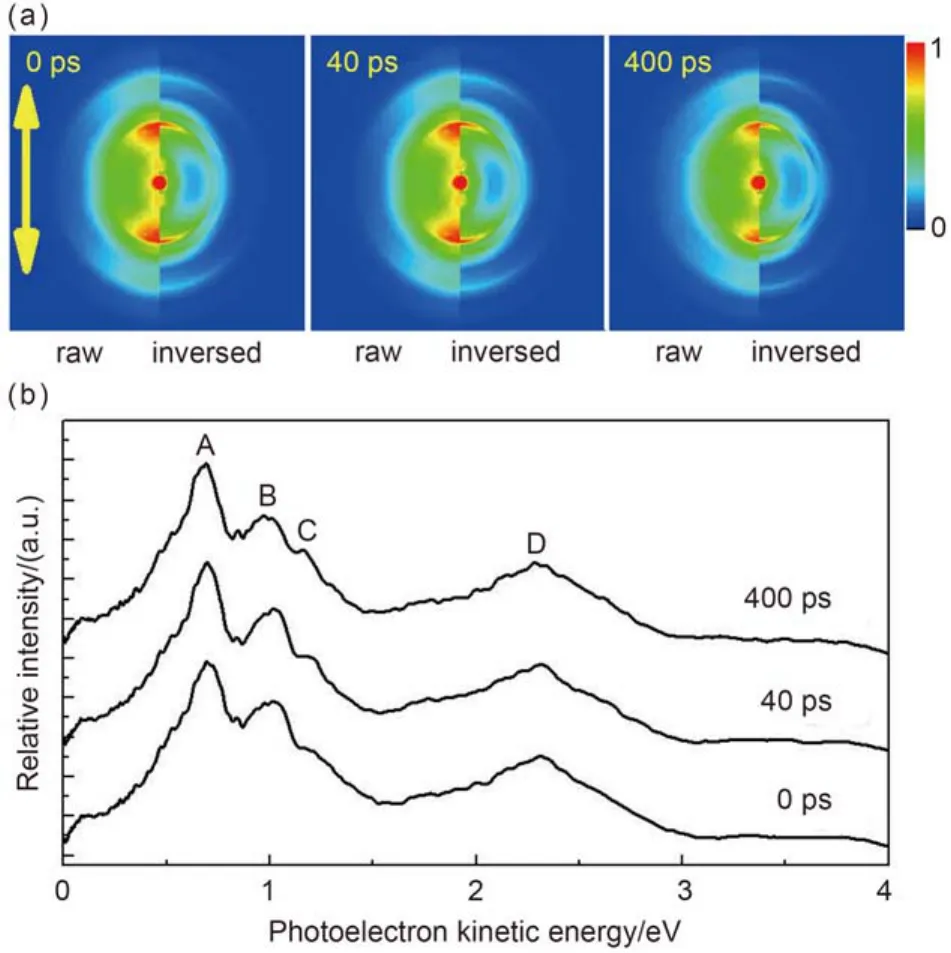
图2 不同延迟时间下S1(1A1)振动基态o-DCB电离产生的光电子图像(a)及相应光电子能谱(b)Fig.2 Photoelectron images produced in ionization of o-DCB at different delay time, excited at the vibrational ground state of the first excited electronic state S1(1A1) (a) and corresponding photoelectron kinetic energy spectra (b)
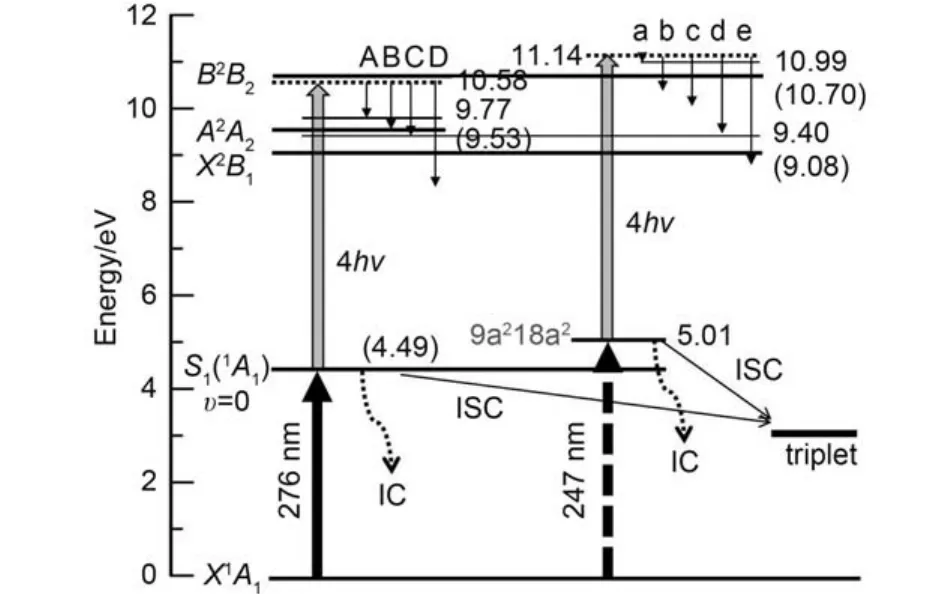
图3 o-DCB电子激发态、离子激发态和相关振动态的能级示意Fig.3 Energy diagram of electronically excited states, ion excited states, and relevant vibrational states of neutral and ionic o-DCB
和o-DCB的S1(1A1)态振动基态激发类似, 由光参量放大器产生的247 nm 激发光带宽约1.6 nm (约260 cm–1), 根据o-DCB双光子光谱实验和理论计算结果,10该激发光可以覆盖的振动模式为微弱的振动模式14118a216b1(40438 cm–1)和14118a2a1(40417 cm–1), 而覆盖不到附近较强的振动模式为14118a2(39973 cm–1). 因此, 该激发波长下无法形成较为有效的相干叠加态.
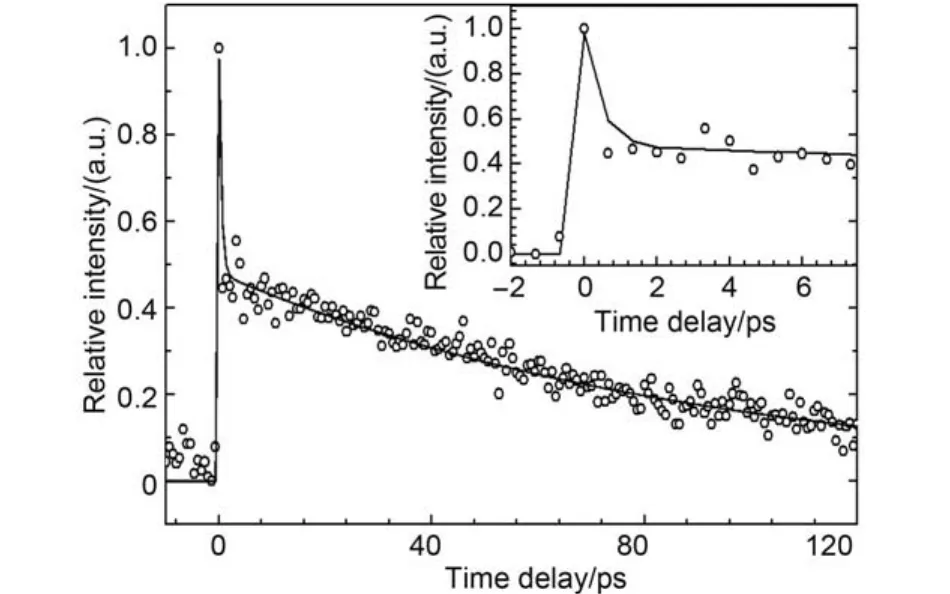
图4 S1(1A1)振动模9a218a2激发时o-DCB母体离子信号强度随延迟时间的变化Fig.4 Delay-time dependence of transient profile of o-DCB parent ion, excited at the vibrational 9a218a2mode of the first excited electronic state S1(1A1)
图5(A)为延时时间为零点时获得的光电子图像(图左半部分)及其重建图像(图右半部分). 图5(B)为相应的光电子能谱, 其中5个独立谱峰能量分别为0.12、 0.73、1.06、1.69和2.31 eV, 图中依次标注为a、b、c、d和e. 这些电子对应的离子态如图3所示.峰a和d分别对应于离子能量为10.992和9.399 eV, 对应的离子态依次为B2B2(00)和X2B1(72101). 峰e在图3中没有对应的下能级. 峰e与b的能量差为1.58 eV, 与814 nm单光子能量(1.528 eV)接近, 因此峰e应当是峰b多吸收一个814 nm光子产生的. 关于峰b和c的来源, 根据2015年Holland等43HeI光电子能谱研究结果, o-DCB在此区域并无明确的离子能级. 峰b和c的能量差为0.33 eV, 对应与阳离子基态及其第一电子激发态的能量之差, 因此峰b和c可能来源于同样的电离通道, 只不过是布居到了不同的阳离子电子态上; 和Holland等43直接从基电子态跃迁到离子态的HeI电离不同, 我们的实验是经过第一电子激发态,电离时离子态和中间态之间的Franck-Condon因子决定了最终电离效率和对应的最大重叠态. Han及其合作者15,21通过量化计算发现o-DCB的单重激发态和三重态对应电子从成键轨道跃迁到反键轨道, 并由此导致C―C键加长. 这种构型上大幅度变化也加强了激发态预解离的速率, 同时也会影响由中间激发态电离产生的光电子能量分布和相对强弱. 峰c对应的阳离子的激发能为0.99 eV, 这也预示着经由中间态的电离过程不能完全用HeI光电子能谱的结果43进行直接比较.

图5 o-DCB的S1(1A1)振动模9a218a2激发时光电子成像(A)及对应的光电子能谱(B)Fig.5 Photoelectron images of o-DCB produced from the vibrational 9a218a2mode of the first excited electronic state S1(1A1) at zero delay time (A) and corresponding photoelectron kinetic energy spectrum (B)
图6是不同延时时间下的光电子能谱. 峰a和d具有几乎相同的衰减趋势, 说明均来自于同一过程,即由高振动激发态向基态高振动激发态的内转换过程. 图6显示峰a和d在400 fs之后就基本上消失了,这也与母体离子双指数模型拟合获得的超快过程((458 ± 12) fs)比较一致. 而峰b、c和e则表现出了较长寿命特征, 对应由S1高振动态向三重态发生的系间窜越过程. 这也是我们不能对峰b和c的振动能确认的一个主要原因, 即可能与三重态有关. 关于o-DCB分子激发态, 特别是三重态的相关信息, 仍需要更多的理论和实验研究.

图6 S1(1A1)振动模9a218a2激发时光电子能谱随延时时间的变化Fig.6 Delay-time dependence of photoelectron kinetic energy spectra of o-DCB produced from the vibrational mode S1(1A1)9a218a2
4 结 论
采用飞秒泵浦-探测技术结合飞行时间质谱和光电子速度成像方法研究了邻二氯苯第一电子单重激发态S1的超快动力学. 用267 nm 波长光将邻二氯苯布居到S1的振动基态, 用814 nm多光子电离检测, 产生的离子态依次为A2A2(v7+ v8)、A2A2(00)和X2B1(72101), 该激发态寿命为(651 ± 10) ps, 对应于S1振动基态向三重态的系间窜越过程. 邻二氯苯处于S1高振动激发态9a218a2时, 观测到了两个衰减通道, 其中一个寿命为(458 ± 12) fs的超快过程对应于S1振动激发态到基电子态的内转换过程; 另一个寿命为(90 ± 10) ps的稍慢过程则对应由S1态振动激发态到三重态跃迁的系间窜越过程. 电离对应的离子态为B2B2(00)和X2B1(72101), 光电子谱中长寿命的谱峰可能与系间窜越过程有关. S1态高振动态的旋轨耦合程度比低振动态的更强, 导致系间窜越过程比振动基态更快.
(1)Zewail, A. H. Angew. Chem. Intl. Edit. Engl. 2000, 39, 2586.
(2)Hertel, I. V.; Radloff, W. Rep. Prog. Phys. 2006, 69, 1897. doi: 10.1088/0034-4885/69/6/R06
(3)Gullett, B.; Oudejans, L.; Touati, A.; Ryan, S.; Tabor, D. J. Mater. Cycles Waste Manage. 2008, 10, 32. doi: 10.1007/s10163-007-0195-8
(4)Gaber, A.; Riese, M.; Grotemeyer, J. J. Phys. Chem. A 2008, 112, 425. doi: 10.1021/jp074802t
(5)Gaber, A.; Riese, M.; Witte, F.; Grotemeyer, J. Phys. Chem. Chem. Phys. 2009, 11, 1628. doi: 10.1039/b816800h
(6)Fujisawa, S.; Oonishi, I.; Masuda, S.; Ohno, K.; Harada, Y. J. Phys. Chem. 1991, 95, 4250. doi: 10.1021/j100164a017
(7)Scharping, H.; Zetzsch, C. J. Mol. Spectrosc. 1995, 112, 8. doi: 10.1016/0022-2852(85)90186-9
(8)Weichhardt, C.; Zimmermann, R.; Schramm, K. W.; Boesl, U.; Schlag, E. W. Rapid Commun. Mass Spectrom. 1994, 8, 381.
(9)Imura, K.; Kishimoto, N.; Ohno, K. J. Phys. Chem. A 2001, 105, 9111. doi: 10.1021/jp011970r
(10)Singh, I. B.; Rai, S. B.; Rai, D. K. J. Mol. Spectrosc. 1994, 163, 364. doi: 10.1006/jmsp.1994.1032
(11)Borg, O. A.; Karlsson, D.; Isomaki-Krondahl, M.; Davidsson, J.; Lunell, S. Chem. Phys. Lett. 2008, 456, 123. doi: 10.1016/j.cplett.2008.03.030
(12)Potts, A. W.; Holland, D. M. P.; Powis, I.; Karlsson, L.; Trofimov, A. B.; Bodzuk, I. L. Chem. Phys. 2013, 415, 84. doi: 10.1016/j.chemphys.2012.12.031
(13)Powis, I.; Trofimov, A. B.; Bodzuk, I. L.; Holland, D. M. P.; Potts, A. W.; Karlsson, L. Chem. Phys. 2013, 415, 291. doi: 10.1016/j.chemphys.2012.09.026
(14)Shimoda, A.; Hikida, T.; Morl, Y. J. Phys. Chem. 1979, 83, 1309. doi: 10.1021/j100473a015
(15)Han, K. L.; He, G. Z. J. Photochem. Photobiol. C-Photochem. Rev. 2007, 8, 55. doi: 10.1016/j.jphotochemrev.2007.03.002
(16)Liu, B. K.; Wang, B. X.; Wang, Y. Q.; Wang, L. Chem. Phys. Lett. 2009, 477, 266. doi: 10.1016/j.cplett.2009.07.025
(17)Liu, Y. J.; Persson, P.; Lunell, S. J. Chem. Phys. 2004, 121, 11000. doi: 10.1063/1.1810135
(18)Sponer, H. Rev. Mod. Phys. 1942, 14, 224. doi: 10.1103/ RevModPhys.14.224
(19)Ichimura, T.; Mori, Y. Chem. Phys. Lett. 1995, 122, 51. doi: 10.1016/0009-2614(85)85476-2
(20)Ichimura, T.; Mori, Y. J. Chem. Phys. 1997, 107, 835. doi: 10.1063/1.474383
(21)Zhu, R. S.; Zhang, H.; Wang, G. J.; Gu, X. B.; Han, K. L.; He, G. Z.; Lou, N. Q. Chem. Phys. 1999, 248, 285.
(22)Szaflarski, D. M.; Simon, J. D.; EI-Sayed, M. A. J. Phys. Chem. 1986, 90, 5050. doi: 10.1021/j100412a035
(23)Yang, J. J.; Simon, J. D.; EI-Sayed, M. A. J. Phys. Chem. 1984, 88, 6091. doi: 10.1021/j150669a005
(24)Diau Eric, W. G.; Casanova, J.; Roberts, J. D.; Zewail, A. H. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 1376. doi: 10.1073/pnas.030524797
(25)Kadi, M.; Davidsson, J.; Tarnovsky, A. N.; Rasmusson, M.; Åkesson, E. Chem. Phys. Lett. 2001, 350, 93. doi: 10.1016/S0009-2614(01)01283-0
(26)Yoshida, N.; Hirakawa, Y.; Imasaka, T. Anal. Chem. 2001, 73, 4417. doi: 10.1021/ac010187s
(27)Deguchi, T.; Takeyasu, N.; Imasaka, T. Appl. Spectrosc. 2002, 56, 1241. doi: 10.1366/000370202760295511
(28)Yuan, L. W.; Zhu, J. Y.; Wang, Y. Q.; Wang, L.; Bai, J. L.; He, G. Z. Chem. Phys. Lett. 2005, 410, 352. doi: 10.1016/j.cplett. 2005.05.103
(29)Yuan, L. W.; Wang, Y. Q.; Wang, L.; Bai, J. L.; He, G. Z. Science in China Ser. B: Chemistry 2004, 47, 283. doi: 10.1360/03yb0251
(30)Suzuki, T. J. Phys. B, At. Mol. Opt. Phys. 2014, 47, 124001. doi: 10.1088/0953-4075/47/12/124001
(31)Liu, Z. M.; Hu, C. L.; Li, S.; Xu, Y. Q.; Wang, Y. M.; Zhang, B. Chem. Phys. Lett. 2015, 619, 44. doi: 10.1016/j.cplett. 2014.11.047
(32)Shen, H.; Chen, J. J.; Zhang, B. J. Phys. Chem. A 2014, 118, 4444. doi: 10.1021/jp500495b
(33)Qiu, X. J.; Ding, Z. H.; Xu, Y. Q.; Wang, Y. M.; Zhang, B. Phys. Rev. A 2014, 89, 033045.
(34)Abulimiti, B.; Zhu, R. S.; Qiu, X. J.; Qin, C.; Zhang, B. Acta Phys. -Chim. Sin. 2014, 30, 22. [布玛利亚阿布力米提, 朱荣淑,邱学军, 秦 晨, 张 冰. 物理化学学报, 2014, 30, 22.]
(35)Liu, Y. Z.; Knopp, G.; Qin, C. C.; Gerber, T. Chem. Phys. 2015, 446, 142. doi: 10.1016/j.chemphys.2014.11.016
(36)Liu, B. K.; Wang, Y. Q.; Wang, L. J. Phys. Chem. A 2012, 116, 111. doi: 10.1021/jp209211s
(37)Parker, D. H.; Eppink, A. T. J. B. J. Chem. Phys. 1997, 107, 2357. doi: 10.1063/1.474624
(38)Eppink, A. T. J. B.; Parker, D. H. Rev. Sci. Instrum. 1997, 68, 3477. doi: 10.1063/1.1148310
(39)Qin, C. C.; Liu, Y. Z.; Zhang, S.; Wang, Y. M.; Tang, Y.; Zhang, B. Phys. Rev. A 2011, 83, 033423. doi: 10.1103/PhysRevA. 83.033423
(40)Garcia, G. A.; Nahon, L.; Powis, I. Rev. Sci. Instrum. 2004, 75, 4989. doi: 10.1063/1.1807578
(41)O'Keeffe, P.; Bolognesi, P.; Coreno, M.; Moise, A.; Richter, R.; Cautero, G.; Stebel, L.; Sergo, R.; Pravica, L.; Ovcharenko, Y.; Avaldi, L. Rev. Sci. Instrum. 2011, 82, 033109. doi: 10.1063/1.3563723
(42)Zakrzewski, V. G.; Ortiz, J. V. J. Phys. Chem. 1996, 100, 13979. doi: 10.1021/jp960978b
(43)Holland, D. M. P.; Powis, I.; Trofimov, A. B.; Bodzuk, I. L.; Soshnikov, D. Y.; Potts, A. W.; Karlsson, L. Chem. Phys. 2015, 448, 61. doi: 10.1016/j.chemphys.2014.11.025
Dynamics of Excited o-Dichlorobenzene
LI Xiao-Ying1WANG Li2,*WANG Yan-Qiu3SONG Zhe1LIU Ben-Kang3,*
(1School of Physics and Electronic Technology, Liaoning Normal University, Dalian 116029, Liaoning Province, P. R. China;2College of Physics and Materials Engineering, Dalian Nationalities University, Dalian 116600, Liaoning Province, P. R. China;3State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning Province, P. R. China)
The dynamics of the first excited singlet electronic state (S1) of o-dichlorobenzene was investigated in real time by the femtosecond pump-probe method combined with time-of-flight mass spectroscopy and the photoelectron velocity mapping technique. The lifetime of the S1vibrational ground state was determined experimentally to be (651 ± 10) ps, corresponding to the intersystem crossing process from the S1state to the triplet state. Two decay channels were found in the S1vibrationally excited mode 9a218a2. The fast process (lifetime constant (458 ± 12) fs) is because of the internal conversion from the S1vibrationally excited mode to the highly vibrationally excited ground state (S0). The slow process (lifetime constant (90 ± 10) ps) is attributed to the intersystem crossing process from the S1state to the triplet state (T1). Photoelectrons with long lifetime characteristics in the spectrum might be connected with the intersystem crossing process. Enhanced spinorbital coupling in the S1highly vibrationally excited state accelerates the intersystem crossing process.
o-Dichlorobenzene; Vibrational excitation; Photoelectron imaging; Photoelectron kinetic energy spectrum
O644
10.3866/PKU.WHXB201506291
Received: April 7, 2015; Revised: June 26, 2015; Published on Web: June 29, 2015.
*Corresponding authors. WANG Li, Email:liwangye@dlnu.edu.cn. LIU Ben-Kang, Email: liubk@dicp.ac.cn.
The project was supported by the National Natural Science Foundation of China (21303199).
国家自然科学基金(21303199)资助项目
© Editorial office of Acta Physico-Chimica Sinica
