五羰基乙酰基锰的深度氢硅烷化
2015-11-30王颖薛冰李艺林李司楠徐崇福
王颖 薛冰 李艺林 李司楠 徐崇福*,
五羰基乙酰基锰的深度氢硅烷化
王颖1,2薛冰1李艺林1李司楠1徐崇福*,1,2
(1常州大学石油化工学院,常州213164)
(2江苏省绿色催化材料与技术重点实验室,常州213164)
采用两步法合成(CO)5MnCOCH3(1),然后对1与Me2PhSiH的深度氢硅烷化反应做9 h的1H NMR连续监控,通过将反应物和衍生物的特征峰与内标物C6H5(CH2)2C6H5的特征峰对照直接积分定量,得到适时含量(mmol)随反应时间改变(h)的变化规律。期间共得到了9种中间体或最终产物,通过核磁波谱对比,确定了这些化合物的分子结构。除了对其中6种化合物的特征峰积分直接定量外,对另外3个化合物采用化学计量结合特征峰积分间接定量,并对其反应机理做出了合理的解释。最后提出了深度氢硅烷化的多步反应与双通道机理,并且对反应原料1和Me2PhSiH中Mn和Si进行了全程跟踪,结果发现,在9 h时,Mn总含量高达90.7%,而Si总含量高达91.4%。
氢硅烷化;积分定量;多步反应;双通道机理
0 引言
早期研究发现[1-2],氢硅烷与具备动力学活性的乙酰基钴及其锰化合物L(CO)3CoCOR(L=CO,PPh3)、(CO)5MnCOR[3-4]反应具有活性。Wegman[5-6]等报道:在过量氢硅烷和一氧化碳存在下,乙酰基化合物与三乙基硅烷在温和条件下发生分子裂解,生成醛类RCHO和相应的金属硅烷基化合物(CO)5MnSiEt3。反应产物可分别通过GC分析和IR光谱检测出。
Gladysz等报道[7-9]:(CO)5MnSiMe3经苯甲醛或乙醛处理后,分离出几种不稳定的α-硅氧烷基化合物。α-三甲基硅氧烷基苄基五羰基锰(CO)5MnCH (OSiMe3)Ph发生Mn-C键均裂分解,生成α-三甲基硅氧烷基联苄和十羰基二锰;而α-三甲基硅氧烷基乙基五羰基锰(CO)5MnCH(OSiMe3)CH3快速发生β-消除,生成五羰基氢化锰(CO)5MnH和三甲基硅氧烷基乙烯CH2=CHOSiMe3。Cutler等研究工作表明[10]:多种氢硅烷均可引发(CO)5MnCOCH3(1)的氢硅烷化反应,外源催化剂的存在仅影响反应诱导期,并不会改变反应的进程。他们还发现1与等物质量的氢硅烷反应定量生成可分离的主产物α-硅氧烷基乙基五羰基锰(CO)5MnCH(OSiR3)CH3和副产物α-硅氧烷基乙烯基五羰基锰(CO)5MnC(OSiR3)=CH2,加入三乙基氢硅烷Et3SiH后,副产物α-硅氧烷基乙烯基五羰基锰(CO)5MnC(OSiR3)=CH2成为主产物。另外,在温和条件下,(CO)5MnCH(OSiR3)CH3与一氧化碳反应,羰基嵌入到Mn-C键之间,生成更加稳定的β-硅氧烷基丙酰基五羰基锰(CO)5MnCOCH(OSiR3)CH3。在过量氢硅烷存在下,(CO)5MnCH(OSiR3)CH3不稳定,会持续降解。在后续反应中,Mn2(CO)10和(R3Si)2O均已得到确认,而作者先前曾预测的几种产物如(CO)5MnC2H5、(CO)5MnSiR3、C2H5OSiR3、[CH3CH(OSiR3)]2,却并没有观察到。
Cutler团队对0.100 mmol 1与0.250 mmol氢硅烷反应得到的产物进行了分离与结构的确认,这对于探讨反应的机理起到关键作用。但其对初始产物(CO)5MnCH(OSiPhMe2)CH3(2)在充足氢硅烷存在下持续降解的探讨,以及一些不明产物确证方面存在一些问题。在本论文后续研究中发现:先前在考察1与Me2PhSiH的1H NMR监控实验中没有确认的δ:0.55(d),是在过量Me2PhSiH存在下,由活性催化剂(Me2PhSi)Mn(CO)4转化成的静置态(Me2PhSi-H) (Me2PhSi)Mn(CO)4(4);而没有确认的δ3.87(m)、δ3.52 (m)、δ3.40(m)和δ1.08(d)处信号则对应于1,2-双(二硅氧烷基)丙烷(Me2PhSiO)CH2CH(OSiPhMe2)CH3(3),则是深度硅烷化主要有机硅产物。通过提供过量的氢硅烷反应物,推动1的深度氢硅烷化。
本文研究了0.100 mmol1与0.350 mmol Me2PhSiH深度硅烷化反应。合成了反应进程中出现的重要化合物,通过比对核磁共振光谱加以确证,并提出了合理的反应机理。
1 实验部分
1.1 主要仪器和试剂
本实验合成操作均使用标准史兰克线,采用仪器包括Perkin-Elmer 1600红外光谱仪,Varian Unity 500核磁共振波谱仪,Hanovia光催化仪、Mbraun Unilab惰性气体手套箱、Buchi真空旋转蒸发仪。以C6D6为溶剂,按化学位移相对于全氘代苯中残余C6H61Hδ:7.15;13Cδ:128.00计算。当1H NMR用于定量测定时,为了消除1H核在不同化学环境下因弛豫时间不同可能引起的积分差异,将脉冲激发信号延时设定为d1=30 s)。在获取29Si核磁光谱时,在C6D6浓溶液中添加Cr(acac)3(0.5mol%),使用反向阀去耦合技术,即90°脉冲和1 s延时。
苯、四氢呋喃、乙醚、己烷等有机溶剂在氩气保护下,从金属钠和二苯甲酮形成的游离基R2CONa+)蓝色溶液中蒸出。氘代溶剂用4A分子筛干燥并且在氩气下储存。液态试剂通过冻结-真空抽吸-融化,循环去除空气并且在氩气下储存,其它固体试剂购回不作处理,直接使用。十羰基二锰Mn2(CO)10购自美国Strem Chemicals公司,三-仲丁基硼氢化钾[CH(CH3)C2H5]3BHK(1 mol·L-1THF溶液)购自美国Acros公司,4-甲氧基氯化苄4-CH3OC6H4CH2Cl、碘甲烷CH3I、氘代苯C6D6、氘代三氯甲烷CCl3D、二甲基苯基氢硅烷(CH3)2(C6H5)SiH、二甲基苯基氯硅烷(CH3)2(C6H5)SiCl、二甲基苯基硅醇(CH3)2(C6H5)SiOH、无水乙醇CH3CH2OH、1,2-丙二醇HOCH2CH(OH) CH3、无水磷酸H3PO4和闪级硅胶(32~57μm)购自美国Aldrich公司。
所有实验过程和波谱数据在supporting information中列出。
1.2 合成
1.2.1 (CO)5MnCOCH3(1)的合成
(CO)5MnCOCH3(1)经两步法合成。按文献[11]的方法,首先以四氢呋喃为溶剂,Mn2(CO)10与[CH(CH3)C2H5]3BHK在低温(-78℃,自然回升至室温)下反应,得到化合物K+[Mn(CO)5]-,然后以无水乙醚为溶剂,K+[Mn(CO)5]-与碘甲烷反应得到甲基五羰基锰CH3Mn(CO)5,升华提纯得到米色晶体,产率为80.9%。最后,在一氧化碳加压条件下,甲基分子内迁徙到邻近羰基上,转化得到浅白色产物1,分离提纯,产率为94.5%。
1.2.2 (CO)5MnCH(OSiPhMe2)CH3(2)的合成
以C6D6为溶剂,1和等物质量Me2PhSiH发生自催化氢硅烷反应,氢硅烷与羰基发生加成反应,产物经分离纯化后得到黄色油状物2,产率为61.0%。
1.2.3 (Me2PhSiO)CH2CH(OSiPhMe2)CH3(3)的合成
与文献[12]中制备Me2PhSiOCH2C(CH3)2CH2OSiPhMe2的步骤类似,以C6D6为溶剂,(CO)5MnBr为催化剂,1.00 mmol 1,2-丙二醇和2.00 mmol Me2PhSiH进行脱氢硅烷化反应,产物经分离纯化后得到无色油状物3,产率为85.0%。
1.2.4 (Me2PhSi-H)(SiPhMe2)Mn(CO)4(4)的原位合成及其含量测定
以C6D6为溶剂,0.100 mmolCH3Mn(CO)5和0.300 mmol Me2PhSiH,在40℃下反应3 h,通过主产物与0.100 mmol内标物质CH3C6H5的特征峰比对计算主产物4的产率,最高积分产率可达75.0%。此外,还检测到CH3CH2OSiPhMe2(10)(特征峰δ:3.57(q,J= 7.0 Hz,2H,OCH2CH3),1.11(t,J=7.0 Hz,3H,CH2CH3),0.33(s,6H,SiCH3)),积分产率为82.0%,其它副产物(CO)5MnSiPhMe2(9)(特征峰δ:0.68(s,6H,CH3)),积分产率为7.0%;(Me2PhSi)2O(7)(特征峰δ:0.31(s,12H,SiCH3)),积分产率为9.0%。
配位不饱和的活性催化剂(Me2PhSi)Mn(CO)4不稳定,无法观察到。但当有过量氢硅烷存在时,通过Si-H与Mn上的配位空缺以抓氢键形成亚稳态的(Me2PhSi-H)(SiPhMe2)Mn(CO)4(4)、过量的氢硅烷以及少量其它副产物的混合物,(特征峰δ:0.55(s,12H,SiCH3),-11.45(s,1H,Mn(Si)H-Si))。分离纯化时,脱离过量氢硅烷后导致4迅速分解。4的C6D6溶液在室温下也会缓慢热分解,同时析出红色三聚体[(CO)4MnH]3。
1.2.5 (CO)5MnSiPhMe2(9)的光化学合成
使用文献报道的光化学合成法[13]。首先,4-ClCH2C6H4CH3与K+[Mn(CO)5]-在乙醚悬浮液中低温(0℃)反应,合成(CO)5Mn-p-CH2C6H4OCH3黄色晶体,产率为92.0%;然后与Me2PhSiH反应,光化学合成目标产物(CO)5MnSiPhMe2,分离纯化得到浅黄色油状物131 mg,产率为79.0%。
1.2.6 (CO)5MnH与Me2PhSiH的反应
首先,K+[Mn(CO)5]-与溶解在C6D6悬浮液中的无水磷酸反应,原位生成(CO)5MnH氘代苯溶液,过滤后用1H NMR化学积分比对内标物质C6H5CH3标定含量为0.042 mmol/600 mg;然后与Me2PhSiH反应,观察到有(CO)5MnSiPhMe2(9)生成。
1.2.7 (CO)5COCH3与Me2PhSiH反应的1H NMR (C6D6)连续观测以及后续处理
将0.100 mmol 1与0.350 mmol Me2PhSiH和0.100 mmol C6H5(CH2)2C6H5(该化合物作为内标物)的C6D6的溶液置于核磁共振仪。1H NMR每隔0.5 h记录一次,连续观察9 h。相对于内标C6H5(CH2)2C6H5特征峰δ:2.73(s,4H,PhCH2)积分。如下信号被用来建立起物料平衡:过量起始原料Me2PhSiH有两处特征峰δ:4.61(s,J=3.7 Hz,1H,SiH)和0.22(d,J=3.7 Hz,6H,SiCH3)。后者信号强度高,谱线简单敏锐,积分效果较好。控制性起始原料1中的δ:2.23(s,3H,MnCOCH3);将首个氢硅烷化产物(CO)5MnCH (OSiPhMe2)CH3(2)中的δ:1.74(d,3H,MnCHCH3),多次硅烷化产物1,2-二硅氧烷基丙烷(Me2PhSiO) CH2CH(OSiPhMe2)CH3(3)中的δ:1.09(d,3H,CHCH3),活性催化剂静置态(Me2PhSi-H)(Me2PhSi)Mn(CO)4(4)中的δ:0.55(s,12H,SiCH3),以及(CO)5MnSiPhMe2(9)中的δ:0.68(s,6H,SiCH3)用作特征峰,共对6个化合物在反应过程中的适时含量(mmol)对反应时间(h)作图,得到反应进程。
当将视窗扩展至高场区时,还观测到三处吸收:1H NMRδ:-7.92(s)、-11.45(s)和-23.18(s)。全部观察结束后,除去溶剂。将红色固体用己烷洗涤后烘干,在史克兰真空线(1.32×10-2Pa)抽吸10 min。用3 mL CH2Cl2溶解,对红色溶液做红外测试。IR:νCO(CH2Cl2)2 015.2、2 012.2、1 995.8 cm-1。再次除去溶剂,真空干燥后得到砖红色固体8 mg。用0.6 mL CDCl3溶解,核磁测试结果:1H NMR(CDCl3)δ:-23.27 (s,1H,MnH)。
2 结果与讨论
2.1 甲基五羰基锰羰化法合成乙酰基五羰基锰
早期文献中,在低温下通过K+[Mn(CO)5]-与乙酰氯的乙酰基化来制备初始产物1[14]。优点是一步即可得到目标产物,缺点是当通过硅胶色谱柱纯化产物时,常伴随部分乙酰基脱去桥羰基,发生自发均裂副反应,得到的最终产物掺杂有(CO)5MnCH3和Mn2(CO)10。由于(CO)5MnCH3和1与氢硅烷的反应通道和产物不同[15],如果起始原料混杂有(CO)5MnCH3,影响最终反应。Mn2(CO)10作为副产物,在后期处理中难以去除。采用两步法合成,首先将K+[Mn(CO)5]-甲基化制得(CO)5MnCH3,经升华纯化后在CO高压作用下,使(CO)5MnCH3羰基化。(CO)5MnCH3升华性强,利用其特性与容易造成污染的副产物Mn2(CO)10分离。(CO)5MnCH3的羰基化是一个干净的“原子经济型”反应,没有副产物生成。
在烷基五羰基锰化合物中羰基的解离或缔合是常态[16]。(CO)5MnCH3在高压CO存在下定量转化为(CO)5MnCOCH3。乙酰基上的羰基来自(CO)5MnCH3配位基团内的邻近羰基。观察到的立体化学表明甲基首先与金属锰脱离,迁移到邻位C=O基团上转化为乙酰基,由此产生的配位空缺随后被外源配体C=O占据[17]。同位素标记研究也证明了这一机理[18]。
2.2 (CO)5MnCOCH3与Me2PhSiH的深度氢硅烷化进程
将由1H NMR积分法直测的(CO)5MnCOCH3(1)、Me2PhSiH、(CO)5MnCH(OSiR3)CH3(2)、Me2PhSiO CH2CH(OSiPhMe2)CH3(3)、(Me2PhSi)2HMn(CO)4(4)和(CO)5MnSiPhMe2(9)6个化合物氢原子核特征峰对照内标C6H5(CH2)2C6H5中4个亚甲基氢原子核积分所获得的适时含量(mmol)为纵坐标,以反应时间(h)为横坐标作图,(CO)5MnCOCH3与Me2PhSiH的深度氢硅烷化反应进程如图1所示。
由图1可知,作为控制性反应物,1的起始量为0.100 mmol,在1 h内耗尽。与此同时,2的含量达到最大值,为0.088 7 mmol。相比之下,Me2PhSiH是过量反应物,起始量为0.350 mmol,在反应过程中不断消耗。它既是构成2的前体,又促进2降解,并形成其它化合物。在9 h时,2仍有0.011 6 mmol的剩余。说明1的深度硅烷化是在有充足Me2PhSiH参与下进行的。发生在金属酰基上的反应是自催化过程,诱导期很短。在过量Me2PhSiH存在下2的降解反应一直在进行。活性催化剂静置态(Me2PhSi-H) (Me2PhSi)Mn(CO)4(4)是在1深度硅烷化过程中产生的一种重要的亚稳态中间体,其含量在5 h时达到最大值0.032 3 mmol,随后缓慢而持续地减少,并伴随着三聚体[(CO)4MnH]3(6)晶体沉淀的生成。另外,化合物(Me2PhSiO)CH2CH(OSiPhMe2)CH3(3)和(CO)5MnSiPhMe2(9)的含量在整个反应进程中持续增加,是1深度氢硅烷化的终极产物,同时3和9的不断生成建立在消耗2以及过量反应物Me2PhSiH的基础上的。
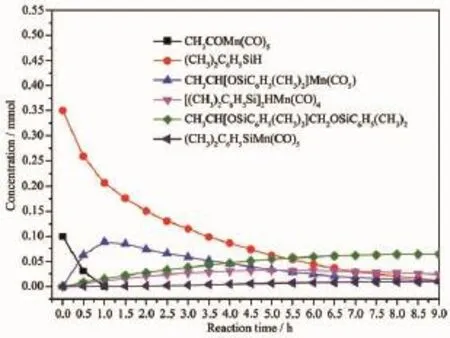
图1 乙酰基五羰基锰与二甲基苯基硅烷的深度氢硅烷化进程Fig.1 Profile of extensive hydrosilation of acetylmanganesepentacarbonyl with dimethylphenylsilane
2.3 反应过程中间物或最终产物的合成及其波谱确定
Cutler等发现[10],在氢硅烷存在下2会持续降解,使用1~2 mmol的氢硅烷,2尚在降解中,氢硅烷已耗尽,反应的后续发展过程终止。本研究试图解决如下几个问题:(1)确定在足量的Me2PhSiH存在下1深度硅烷化反应结果。(2)对前人曾观察到,但尚未得到确证的1H NMR信号所代表的化合物进行表征,追踪起始反应物中硅元素和锰元素的归宿,理清反应链中各化合物的相互关系。(3)探讨1与Me2PhSiH深度氢硅烷化反应的反应机理。为了提高光谱数据的可靠性,合成了在1与Me2PhSiH反应过程中1H NMR所观察到的分子结构相同的化合物。选择适当的特征峰直接定量,或在定性后,再比对机理分析和化学计量进行量化处理,使结论从质到量都尽可能贴近实际情况。
2按照文献方法合成[19],1与等物质量的Me2PhSiH反应生成主产物2,重复了Cutler小组的研究结果,但(CO)5MnC(OSiMe2Ph)=CH2浓度极低,可以忽略不计。这与只选用Et3SiH时才会得到较高含量α-硅氧烷基乙烯基五羰基锰的结论一致[20]。与Et3SiH相比,Me2PhSiH本就不利于α-硅氧烷基乙烯基化合物的形成,而过量的Me2PhSiH存在下,进一步抑制α-硅氧烷基乙烯基化合物产生[21]。另外,在过量Me2PhSiH(0.350 mmol)的体系中,反应链进一步延长,2仅仅是一个过渡化合物,在反应初期大量形成,达到峰值后逐渐降解,转化成多个衍生物。选择δ:1.74(d,3H,MnCHCH3)作为2的特征峰,是基于附近没有别的吸收,谱线相对简单,且积分强度适中。3是在过量Me2PhSiH存在下,由2降解所形成的最主要有机硅产物。Cutler等没有验证该化合物1H NMR谱图,认为其是2,3-双二甲基苯基硅氧基丁烷CH3CH(OSiPhMe2)CH(OSiPhMe2)CH3,直到合成了该化合物,比对核磁波谱后发现与先前实验中出现的谱图不吻合后排除[22]。合成并确证了3的结构,解决了1与Me2PhSiH深度硅烷化中的一个主要难题。作为外消旋和非对咉体混合物,在分子3的1H NMR波谱中,C1和C2上的氢原子展现出独特的模式δ:3.56~3.53,3.42~3.38(m,2H,CH2),3.89~3.88(m,1H,CH)。而C3上的氢原子则展现普通的模式δ:1.09(d,J=6.1 Hz,3H,CHCH3)。此处信号简洁,强度适中,附近没有别的干扰,因此,以此作为特征峰计算3的含量。活性催化剂R3SiMn(CO)4的观念早就被提出[23],然而这个配位不饱和化合物因其高度动力学活性而难以捕捉。它的光谱特征尚未得到确认。在过量氢硅烷存在下,Si-H键的σ电子云填补Mn原子空缺配位点,形成σ配位键(抓氢键),使处于静置态的活性配合物在一定程度上得以稳定。在本实验中通过热化学反应原位合成,并表征了这个亚稳态化合物4,1H NMR积分产率最高达75.0%。在特征峰1H NMRδ:0.55(s,12H,SiCH3)附近没有其它信号重叠,可用于准确量化分析。在高场呈现的化学位移1H NMR(C6D6)δ:-11.45(s,1H,MnHSi2)是4存在的另一个特征数据,电负性小于氢原子的Mn原子和Si原子束缚电子云的能力均较弱,无论是Mn-H键还是Si-H键,电子云都偏向氢原子,因此,该化合物中与金属锰链接的H核被高度屏蔽,其化学位移处于高场为负值。Me2PhSi-SiPhMe2(5)的合成是普通的伍兹偶合,由于它的特征峰1H NMR(C6D6)δ:0.29(s,12H,SiCH3)与Me2PhSi-O-SiPhMe2(7)的特征峰1H NMR(C6D6)δ:0.31(s,12H, SiCH3)极其靠近,部分重叠,在积分时难以正确把握拐点。因此,确定5的含量须从在δ:0.29~0.44区间总积分中扣除2、3、7相关积分值间接获得。三聚体6的合成过程包括在氢硅烷诱导下,锰配位圈中甲基向邻近羰基上的迁徙、乙酰基上的氢硅烷加成、氢化裂解、寡聚4个步骤。另外6的IR波谱呈现4个吸收峰,与C2v点群,2A1+B1+B2红外活性吸收模式相吻合。特征1H NMR高场吸收δ:-23.27(s,3H,Mn-H-Mn)是处于2个Mn原子之间的桥基氢原子核被高度屏蔽的证据。
(Me2PhSi)2O(7)是Me2PhSiH热化学过程中经常出现的附带产物[24]。按照文献中有关醇的脱氢硅烷化反应条件,在(CO)5MnBr催化下,使用Me2PhSiOH和Me2PhSiH便捷合成该化合物。(CO)5MnH(8)的合成具有挑战性。因K+[(CO)5Mn]-对水分子高度敏感,使用固体无水磷酸作为氢源。另外,化合物8挥发性极强,无法从乙醚等低沸点溶剂中直接分离。在C6D6中原位合成这个化合物,不经分离直接使用。8的IR波谱出现3个吸收峰,与该化合物属于C4v点群,具有2A1+E红外活性模式相吻合。1H NMR(C6D6) δ:-7.92(s,1H,Mn H),是该化合物的另一化学特征峰,表明氢原子核被高度屏蔽。(CO)5MnSiPhMe2(9)是Me2PhSiH与(CO)5MnCOCH3深度氢硅烷化中出现的关键化合物之一。同使用卤代烷RX与K+[(CO)5Mn]-的金属烷基化制备(CO)5MnR情况不同,卤代硅烷与五羰基锰钾盐的反应不稳定,成功率较低[25]。本文采用光化学合成方法便捷高效地得到了9的高纯样品[26]。
2.4 (CO)5MnCOCH3与Me2PhSiH的深度氢硅烷化反应机理
由1H NMR直接测定了6个重要化合物含量随时间的变化规律,得到相关反应通道的重要信息。完整的反应线路图不仅包括“显性”化合物,还包括“隐性”化合物[27],即在1H NMR谱图中没有出现特征峰,或因特征峰附近存在信号重叠,分辨不好而没能准确检测出的潜在化合物。一些存在时间过短的中间过渡态未达到能够检测出的浓度,但却是构成数据链的关键环节,也必须考虑在内,用符号T标示。根据对整个反应进程中的数据分析,推断反应机理如图2所示。
首先,Me2PhSiH与控制性反应物1上的羰基发生加成反应,转化为2;α-硅氧烷基乙基迁移至Mn配位圈中邻近的羰基上,产生β-硅氧烷基丙酰基瞬时中间体T1,分子内迁移产生的配位空缺立即被游离态Me2PhSiH分子的Si-H键σ电子云填补,形成抓氢键配合物,起到稳定作用。T1与Me2PhSiH再次发生加成反应,转化为α,β-双硅氧烷基丙基瞬时中间体T2。然后与Me2PhSiH反应,导致Mn-C键和Si-H键的同步断裂,并交叉偶合产生3和4。理论上,在4中2个硅烷基与金属锰原子之间的成键模式不同,1个是普通Mn-Si的σ键,而另1个是Si-H σ-电子云与Mn之间的抓氢键。而事实是在4分子内Me2PhSi-H,Me2PhSi以及与游离态的Me2PhSiH处于不断的交换之中。观察到的1H NMRδ:0.55(s,12H,MnSiCH3)只有1种吸收,并没有分裂成二重峰,说明2个硅烷基在1H NMR时间框架中并没有区别。发生在4与氢硅烷之间的交换应该是快速且可逆的,且只有过量氢硅烷存在才能维持4稳定。悬蒸除去溶剂,制备无溶剂的4时,低沸点的Me2PhSiH随溶剂蒸出。一旦脱离过量Me2PhSiH,4立即分解。4与游离氢硅烷保持着极不稳定的动态平衡状态。但是,如果从4中解离出来的不是Me2PhSiH而是Me2PhSi-SiPhMe2(5),情况就大不相同。4能够在过量Me2PhSiH中较长时间稳定存在,说明解离出5要比解离出Me2PhSiH困难得多。然而,实际情况是4的确缓慢而又持久地降解,说明解离出5的过程虽不迅速却一直进行,一旦4中的2个硅烷基之间发生分子内偶合,形成硅烷二聚体(Me2PhSi)2(5),反应就不可逆转。当5从4中解离后,留下配位不饱和瞬时中间体T3。体系中存在的过量氢硅烷虽有可能通过抓氢键填补T3的空位,生成类似活性中间体静置态(Me2PhSi-H)HMn(CO)4,但该化合物远不及4稳定。最终通过寡聚化,形成配位饱和,难溶三聚体6,从溶液中析出。化学惰性的5和3以及低溶解度的6作为终极产物群驱使2和4的降解化学平衡不可逆转地向右移动,也是其不能够长期保持稳定的原因。

图2 乙酰基五羰基锰与二甲基苯基硅烷的深度氢硅烷化机理Fig.2 Proposed mechanism for the extensive hydrosilation of acetylmanganesepentacarbonyl with dimethylphenylsilane
反应过程中还检测到Me2PhSiMn(CO)5(9),含量不高,最多时仅有9.2%。在追踪其生成机理时存在迷失环节。鉴于该化合物中的存在5个端羰基,只能由处在反应链上游的1或2反应生成。在这个无外源C=O补充的体系中,一旦发生了烷基的分子内迁移活动,将1个端羰基转换成了酰基就不可能得到五羰基产物了。9不可能直接来自1,如果是1与Me2PhSiH反应导致了Mn-C键断裂,其产物除了9之外,还应当有伴生产物乙醛。但实验中并没有检测到乙醛。另外,乙醛与Me2PhSiH发生了羰基上的加成反应,必然转化为CH3CH2OSiPhMe2(10),但实验中也没有检测到这个化合物。况且1在反应不到1 h即消耗殆尽,而9则是自始至终一直增加。因此,可以完全排除9来自1的可能性。2成为它唯一的来源。如果2与Me2PhSiH直接反应导致Mn-C键断裂,除了生成9之外,另1个伴生产物依然是CH3CH2OSiPhMe2,这也与事实不符。因此,9来自1或2与Me2PhSiH直接裂解氢硅烷化可以完全排除。对9的来源可做如下推理:2在氢硅烷诱导下的降解反应除了按1个主要通道进行,产生化合物3、4、5、6外,还按另1个次要通道进行,产生化合物7、8、9。即2与Me2PhSiH进行脱氧氢硅烷化反应,产生(Me2PhSi)2O(7)和(CO)5MnCH2CH3,7确实被检测到,而(CO)5MnCH2CH31H NMRδ:1.28(t,J=7.1 Hz,3H,CH3),1.13(q,J=7.1 Hz,2H,CH2)并没有检测到。考虑到过渡金属乙基化合物极具动力学活性。β-碳上的氢原子会自发脱离配体迁徙到金属中心,形成氢化物,如果发生这种情况,配位乙基因失去1个β-氢原子,会以乙烯气体分子形式消去。因此在这个有大量活性催化化合物存在的体系中,(CO)5MnCH2CH3的性质很不稳定,因此没有被1H NMR检测出。普通NMR试管橡皮塞无法阻止气体逸出,即使产生少量乙烯伴生产物,也会立即脱离液相,因此没有在液相中形成足够分量被观察到。反应过程中在高场观测到的1H NMRδ-7.92微弱信号,可以作为(CO)5MnH(8)存在的证据。实验证实:(CO)5MnH与Me2PhSiH反应产物为(CO)5MnSiPhMe2(9)。从1H NMR确实观察到化合物9,而它潜在的伴生产物H2气体,从溶液中逃逸,并没能被观测到。因此,2发生脱氧硅烷化产生7和(CO)5MnCH2CH3,而(CO)5MnCH2CH3的β-氢消除产生(CO)5MnH(8),接下来8与体系中过量Me2PhSiH脱氢硅烷化产生9。在2通过次要通道降解的反应中,伴随着气体放出,促使反应平衡不可逆地进行。
2.5 (CO)5MnCOCH3与Me2PhSiH的深度氢硅烷化中的物料平衡
通过1H NMR中特征峰对比可计算6个化合物的适时含量,从而得到含量随时间的变化曲线图,结合理论推测可知,间接量化另外3个化合物做补充,提出了反应机理。上述结论的合理性还需要通过物料衡算加以验证。5的特征峰δ:0.29(s,12H,SiCH3)与其它几个含有硅甲基的化合物在这个吸收区间靠的太近,谱线部分重叠。如2δ:0.33(s,6H,SiCH3);3δ:0.44,0.35(s,12H,SiCH3);7δ:0.31(s,12H,SiCH3)。然而2的含量可从δ:1.74(d,3H,MnCHCH3)处,3的含量可从δ:1.09(d,3H,CHCH3)精准测定。和5的情况类似,7的含量也无法通过1H NMR积分准确测定,从机理分析得知,7是促使9生成的伴生化合物,并且化学性质稳定。动力学活性的化合物(CO)5MnCH2CH3因极易发生β-消除而无法稳定存在体系中,但它的衍生化合物8在过量Me2PhSiH存在下会定量转化为9。因此,在中间物无显著积累情况下,可以认为7和9的含量基本相同,该化合物含量有限,在反应结束时才达到最大值0.092 mmol,数值误差对于建立反应进程的影响不大,由于9可以不受干扰地从δ:0.68(s,6H,CH3)准确测定。因此,在2、3和7都能准确测定的情况下,从δ:0.29~0.44的积分总量中,扣除在此区间内几个已知化合物积分之和,剩余部分就是5的积分。在三聚物6的1H NMR(C6D6)跟踪中,虽可以从-23.27(s)处的微量吸收对它定性,但因大部分6以晶体析出脱离液相而无法定量。按照反应机理和化学计量推测,6的含量应当是它的先驱体,即瞬时过渡态T3含量的三分之一,而T3作为5的伴生化合物,应当与5取相同值,所以6含量应当是5含量的三分之一。经过9 h,在反应检测结束时,5的含量为0.046 0 mmol,按照机理推测,6的含量就应当是0.153 mmol。在处理用于研究反应进程试样沉淀物时,曾分离出8 mg红色固体,经IR表征与6的物化特征吻合。分离含量经换算相当于0.015 9 mmol,实测结果与推测分析基本一致。
在追踪反应体系中Si源及Mn源归宿时发现:如果只将直接观测到的有机锰化合物中锰含量数值进行求和,即∑n(Mn)=n1(Mn)+n2(Mn)+n4(Mn)+n9(Mn),直接测量Mn总量随着反应进程直线下降,到9 h时仅为0.044 7 mmol,即只能指认44.7%Mn源的下落;而如果连同推测的“隐性”Mn源包括在内,即∑n(Mn)= n1(Mn)+n2(Mn)+n4(Mn)+n9(Mn)+3n6(Mn),Mn总量随时间变化不大,即使在9 h时,仍能指认90.7%以上Mn源的下落。与Mn源的情况类似,如果只将直接测量的有机硅化合物相加,即∑n(Si)=nMe2PhSiH(Si)+n2(Si)+2n3(Si)+2n4(Si)+ 2n5(Si)+n9(Si),直接测量Si总量也随时间有较大下降,在9 h时,也仅能指认59.9%Si源的下落;如果将根据机理推测和化学计量法综合定量的7也包括在内,即∑n(Si)=nMe2PhSiH(Si)+n2(Si)+2n3(Si)+2n4(Si)+2n5(Si)+n9(Si)+ n7(Si),Si总量保持相对平稳,在9 h时仍能指认91.4%以上的Si源。反应过程中的物料衡算在表1中列出。
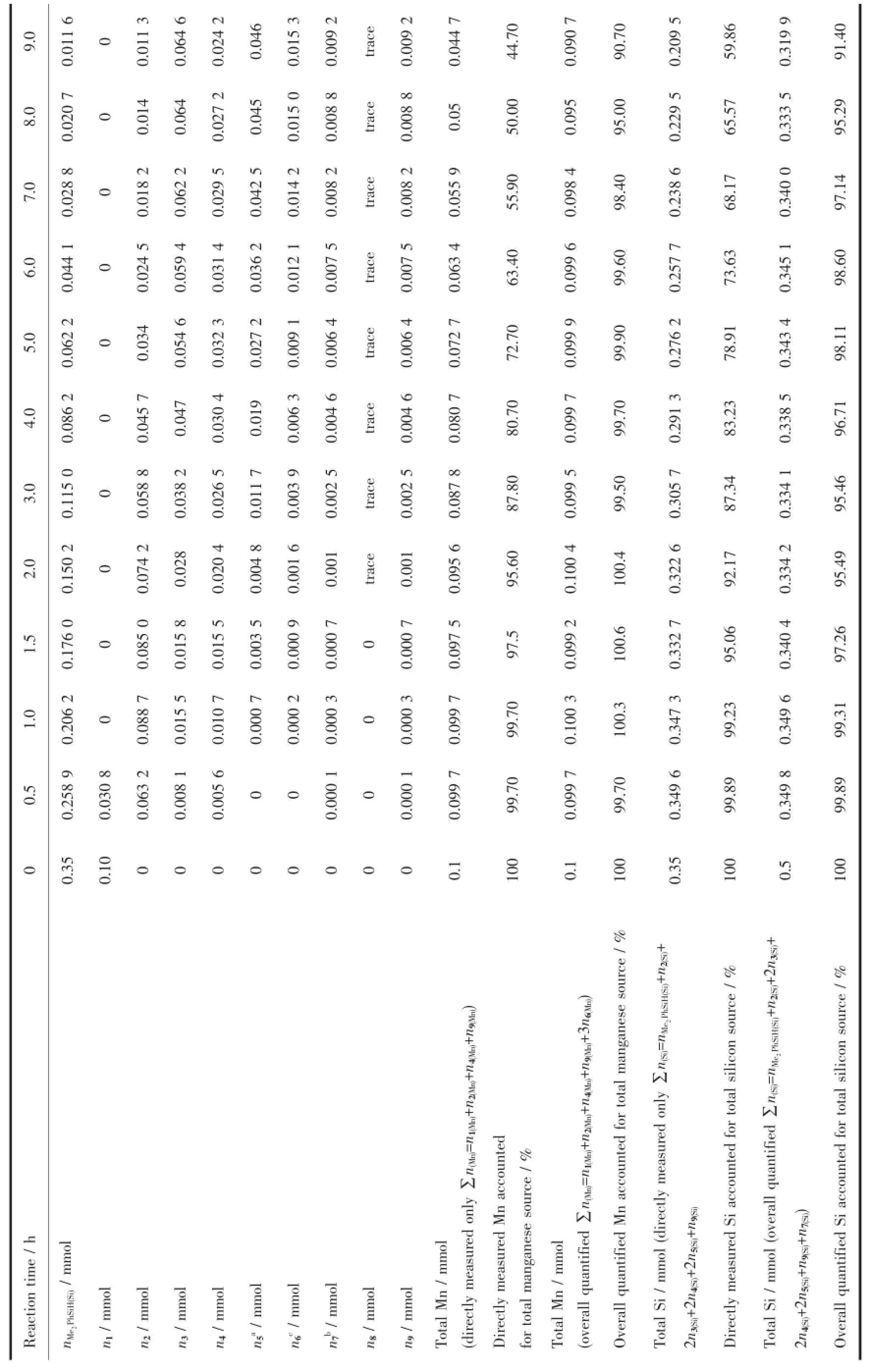
表1 (CO)5MnCOCH3与Me2PhSiH 的深度氢硅烷化反应进程物料平衡Table 1 Material balance for the extensive hydrosilation process of acetyl manganese pentacarbonyl with dimethylphenylsilane
3 结论
本文采用两步法合成(CO)5MnCOCH3。首先,通过CH3I与K+[Mn(CO)5]-的甲基化反应制取(CO)5MnCH3,升华纯化去除Mn2(CO)10等杂质后,在高压CO气氛中羰基化得到目标产物(CO)5MnCOCH3。
对(CO)5MnCOCH3与Me2PhSiH的C6D6反应液做了9 h1H NMR连续监控,将反应原料Me2PhSiH2、(CO)5MnCOCH3以及反应中间体,衍生物(CO)5MnCH (OSiPhMe2)CH3、Me2PhSiOCH2CH(OSiPhMe2)CH3、(Me2PhSi-H)(Me2PhSi)Mn(CO)4、(CO)5MnSiPhMe2的特征峰对照内标物C6H5(CH2)2C6H5的特征峰直接积分得到的适时含量(mmol)为纵坐标,反应时间(h)为横坐标作图,获得9 h反应进程。
制备了9个在(CO)5MnCOCH3与Me2PhSiH深度硅烷化中出现的中间体或最终衍生产物,通过真实样品谱图与反应进程谱图比对,对反应过程中化合物加以确认或排除。
提出了(CO)5MnCOCH3与Me2PhSiH深度硅烷化进程中多步反应机理和双通道机理。
对2种起始反应原料(CO)5MnCOCH3和Me2PhSiH中Mn源和Si源进行全程跟踪。9 h时,Mn总含量仍然高于90.7%,Si总含量高达91.4%。
[1]Gregg B T,Hanna P K,Crawford E J,et al.J.Am.Chem. Soc.,1991,113(47):384-385
[2]Hanna P K,Gregg B T,CutlerA R.Organometallics,1991, 10(1):31-33
[3]Ziegler T,Verslius L,Tschinke V J.J.Am.Chem.Soc., 1986,108(21):612-614
[4]Axe F U,Marynick D S.Organometallics,1987,6(17):572-575
[5]Wegman R W.Organometallics,1986,5(3):707-710
[6]Kovacs I,Sisak A,Ungvary F,et al.Organometallics,1988,7 (10):1025-1029
[7]Glaysz J A.Acc.Chem.Res.,1984,17(57):326-330
[8]Selover J C,Vaughn G D,Strouse C E,et al.J.Am.Chem. Soc.,1986,108(11):1455-1457
[9]Vaughn G D,Glaysz J A.J.Am.Chem.Soc.,1986,108(35): 1473-1475
[10]Gregg B T,Cutler A R.J.Am.Chem.Soc.,1996,118,42 (19):10069-10084
[11]Krein K A,Gladysz J A.Organometallics,1986,5(15):936-940
[12]Mars M,Brinkman K C,Lisensky C A,et al.J.Org.Chem., 1985,50(24):3396-3398
[13]XU Chong-Fu(徐崇福),FANG Jun-Zhuo(房俊卓),CHEN Miao(陈苗),et al.Acta Chim.Sin.(化学学报),2008,66(10): 1239-1244
[14]Gregg B T,Cutler A R.Organometallics,1994,13(9):1039-1043
[15]XU Chong-Fu(徐崇福),FANG Jun-Zhuo(房俊卓),XUE Bing (薛冰),et al.Acta Chim.Sin.(化学学报),2011,69(8):999-1006
[16]Gladysz J A,Williams G M,Tam W,et al.Inorg.Chem., 1979,18(3):553-558
[17]Warner K E,Norton J R.Organometallics,1985,4(16):2150-2154
[18]Tan K Y D,Teng G F,Fan W Y.Organometallics,2011,30 (15):4136-4143
[19]Anderson G K.Acc.Chem.Res.,1984,17(21):67-74
[20]Gregg B T,Cutler A R.Organometallics,1998,17(32):4169-4175
[21]Gray L M,Donald A T.Inorganic Chemistry.New Jersey: Prentice Hall,1999:297
[22]Gregg B T,Cutler A R.J.Am.Chem.Soc.,1995,117(10): 10139-10140
[23]Mao Z,Gregg B T,Cutler A R.Organometallics,1998,17(10): 1993-2002
[24]Green M L H,Nagy P L I.J.Organomet.Chem.,1963,1(27): 58-60
[25]Xu C F,Anderson G K A.Organometallics,1996,15(7):1760-1764
[26]XU Chong-Fu(徐崇福),FANG Jun-Zhuo(房俊卓),XUE Bing (薛冰),et al.Acta Chim.Sin.(化学学报),2009,67(20):2355-2362
[27]LI Rong(厉荣),CHEN Peng-Gang(陈鹏刚).Chinese J. Inorg.Chem.(无机化学学报),2008,24(20):657-660
Extensive Hydrosilation of Acetyl Manganese Pentacarbonyl
WANG Ying1,2XUE Bing1LI Yi-Lin1LISi-Nan1XU Chong-Fu*,1,2
(1College of Petrochemical Engineering of Changzhou University,Changzhou,Jiangsu 213164,China)
(2Jiangsu Province Key Laboratory of Green Catalytic Materials and Technologies Changzhou,Jiangsu 213164,China)
(CO)5MnCOCH3(1)was synthesized in two steps.A solution 1 with Me2PhSiH in C6D6was continuously monitored by1H NMR spectroscopy for 9 h and reaction profile was established using timely contents(mmol) obtained by directly integrating chemical labels of the reactants and derivatives against that of the internal standard C6H5(CH2)2C6H5.9 intermediates and final products emerged in the extensive hydrosilation were prepared and the molecular structures of the derivations generated in the reaction process were positively confirmed by comparison of their NMR spectra with that of real substances.All besides the 6 species that were quantified by direct integration of their chemical labels the other 3 were indirectly quantified by combination of chemical stoichiometry and integration of their chemical labels.Attempt was made to make plausible explanations for the origins of these species.Finally,a mechanism with multiple step reactions process and double pathways was proposed.More than 90.7%of the total manganese source was accounted for at 9 h and meanwhile that of more than 91.4%ofthe totalsilicon source was accounted for.
hydrosilation;integrating chemical labels;multiple step process;double pathways mechanism
O643.32
A
1001-4861(2015)07-1393-09
10.11862/CJIC.2015.194
2015-03-23。收修改稿日期:2015-05-22。
国家自然科学基金(No.21376032)资助项目。
*通讯联系人。E-mail:xu_chf@163.com,Tel:13961224298
