基于双齿咪唑席夫碱配体的两个手性自旋转换铁(Ⅱ)配合物
2015-11-30顾玲任冬红刘志明孙晓利邱丹顾志国李在均
顾玲 任冬红 刘志明 孙晓利 邱丹 顾志国*,,2 李在均
基于双齿咪唑席夫碱配体的两个手性自旋转换铁(Ⅱ)配合物
顾玲1任冬红1刘志明1孙晓利1邱丹1顾志国*,1,2李在均1
(1江南大学化学与材料工程学院,无锡214122)
(2食品胶体与生物技术教育部重点实验室,无锡214122)
以含有苯环和咪唑环的手性双齿席夫碱为配体,合成了2个纯手性单核自旋转换铁(Ⅱ)配合物fac-Δ-[Fe(S-L1)3][ClO4]2(1),mer-Λ-[Fe(R-L2)3][ClO4]2·Et2O(2)(L1=1-对氯苯基-N-(1-正丙烯基-1H-咪唑-2-亚甲基)乙胺;L2=1-苯基-N-(1-异丙烯基-1H-咪唑-2-亚甲基)乙胺)。利用X-射线单晶衍射、元素分析(EA)、红外光谱(IR)、核磁共振氢谱(1H NMR)、紫外光谱(UV)、圆二光谱(CD)等手段对配合物结构进行了表征。X-射线单晶衍射表明在配合物1和2中,铁(Ⅱ)金属中心与3个不对称双齿手性席夫碱配体中的6个氮原子配位形成八面体配位环境。配合物1中每个结构基元中包含1个[Fe(Ln)3]2+阳离子和2个高氯酸根阴离子。而配合物2中每个结构基元中包含2个[Fe(L n)3]2+阳离子、4个高氯酸根阴离子和1个乙醚分子。由于铁(Ⅱ)中心周围手性配体的螺旋协调配位使[Fe(L n)3]2+形成单一构型。Fe(Ⅱ)-N键长表明配合物1中的铁(Ⅱ)在低自旋状态,而配合物2中的铁(Ⅱ)在高自旋状态。在[Fe(L n)3]2+中,相邻配体中的苯环和咪唑环形成分子内π-π相互作用。配合物1和2通过分子间C-H…π和C-Cl…π相互作用形成超分子结构。CD光谱证实配合物1和2在溶液中的光学活性。磁性测试表明配合物1和2分别在372 K和146 K发生自旋转换。由于配合物1和2具有不同的堆积方式和分子间相互作用,导致1和2表现出不同的自旋转换温度。
纯手性;自旋转换;铁(Ⅱ)配合物;席夫碱
0 引言
某些含有3dn(4≤n≤7)的过渡金属配合物在一个适当的外干扰下(如:温度、压力、光辐射等)能发生自旋转换(即高自旋态和低自旋态),这种磁双稳态现象由于在信息存储、温度开光和传感等领域的潜在应用而越来越受到关注[1-3]。手性自旋转换配合物能够提供潜在的新性质,比如磁手性二向色性和铁电性等[4-8]。为了开发手性自旋转换分子材料,我们将研究集中在基于光学纯咪唑席夫碱为配体的三(二亚胺)铁(Ⅱ)化合物上。如图1(a)所示,当合成1个含有3个不对称双齿配体的三螯合化八面体配合物时,会形成mer构型和fac构型2种几何异构体,nmer∶nfac趋向于3∶1(图1a)。此外,三螯合化八面体金属中心还存在对应异构体Δ和Λ构型(图1a),从而导致fac-Δ,fac-Λ,mer-Δ和mer-Λ共存的复杂异构问题[9-10]。由于拆分这4种构型配合物既费力又低效益,因而简单有效地合成单一构型八面体配合物成为长久的挑战[11-13]。当采用纯手性配体时,如果邻近的手性基团之间有选择性相互作用,手性就能够扩展到更高范围,发生配体到金属中心的手性传递,这样就可能得到单一构型的三螯合配合物[14-17]。为了得到期望的纯手性自旋转换配合物,将带有苯环和咪唑环的手性二齿席夫碱配体引入到金属化合物中,此类含咪唑双齿席夫碱配体能够为自旋转换的发生提供适当的配体场。此外,手性席夫碱配体在金属配合物形成过程中起到构型诱导作用,通过配位、π…π和C-H…π相互作用协同控制金属中心的最终构型[18-20]。
在本文中,我们通过简单的多组分自组装方法一步合成了2个纯手性铁(Ⅱ)自旋转换配合物。如图1(b)所示,通过改变2个能够影响自旋转换行为的1-咪唑取代(R1)组分和4-苯乙胺取代(R2)组分,成功合成了含手性咪唑席夫碱配体Ln(n=1,2)的2个手性单核亚铁配合物,并通过X-射线单晶衍射、元素分析(EA)、红外光谱(IR)、核磁共振氢谱(1H NMR)、紫外光谱(UV)、圆二光谱(CD)等手段对配合物结构进行了表征,并对其自旋转换性能进行了研究。

图1 (a)八面体配合物的手性构型,(b)配合物1和2的合成路线Fig.1(a)Chiral configurations of octahedral complexes;(b)Preparation route of complexes 1 and 2
1 实验部分
1.1 仪器与试剂
FTLA2000型傅立叶红外光谱仪,扫描范围4 000~500 cm-1;AVANCEⅢ400MHz全数字化核磁共振谱仪;Elementar Corporation Vario ELⅢ元素分析仪;TU1900型双光束紫外可见分光光度计;MOS-450/AF-CD圆二色谱仪;MPMS-XL-7超导量子干涉仪磁性测量系统;APEXⅡDUO CCD衍射仪;1-甲基-1H-咪唑-2-甲醛;3-溴丙烯;3-溴-2-甲基丙烯;(R)-(+)-α-甲基苄胺;(S)-(-)-对氯苯乙胺;六水合高氯酸亚铁;N,N-二甲基甲酰胺;乙腈;无水乙醚;所用试剂均为分析纯。
1.2 合成
1.2.1 3-(咪唑-2-甲醛)丙烯和2-甲基-3-(咪唑-2-甲醛)丙烯的合成
称取10 mmol 1-甲基-1H-咪唑-2-甲醛(0.96 g)、12 mmol烯丙基溴(1.45 g)或12 mmol3-溴-2-甲基丙烯(1.45 g)、12 mmol无水碳酸钾(1.65 g),加入到20 mL N,N-二甲基甲酰胺中,此反应液在氮气保护下50℃搅拌3 d。冷却过滤,滤液加10 mL水并用40 mL乙酸乙酯萃取4次,收集有机层并用饱和氯化钾水溶液洗涤,接着用无水硫酸镁干燥,最后旋转蒸发去除溶剂。所得粗产品在40℃真空干燥箱中干燥得到黄色油状液体。3-(咪唑-2-甲醛)丙烯的产率:59%。IR(KBr,ν/cm-1):3 117,2 845,1 684,1 478,1 406,1 336,765。1HNMR(400 MHz:CD3CN,δ):9.704 (s,1H1),7.330(s,2H2),7.25(s,2H3),6.023(m,1H5),5.197 (d,J=2.3 Hz,2H4),5.008(d,J=1.7 Hz,2H4)。C7H8N2O的元素分析计算值(%):C 61.75;H 5.92;N 20.58;实验值(%):C 61.70;H 5.90;N 20.54。2-甲基-3-(咪唑-2-甲醛)丙烯的产率:65%。IR(KBr,ν/cm-1):3 114,2 986, 2 935,2 859,1 694,1 470,1 412,1 335,1 169,766。1H NMR(400 MHz:CD3CN,δ):9.704(s,1H1),7.310(s,2H2),7.262(s,2H3),4.949(s,2H5),4.461(s,2H4),1.700 (s,3H5)。C8H10N2O元素分析计算值(%):C 63.98;H 6.71;N 18.65;实验值(%):C 63.92;H 6.67;N 18.61。
1.2.2 配合物1和2的合成
称取0.3 mmol(S)-(-)-对氯苯乙胺(0.046 8 g),0.3 mmol 3-(咪唑-2-甲醛)丙烯(0.447 g)或0.3 mmol (R)-(+)-α-甲基苄胺(0.366 g),0.3 mmol 2-甲基-3-(咪唑-2-甲醛)丙烯,加入10 mL乙腈溶剂,此反应液在氮气保护下80℃下搅拌回流2 h。反应充分完成后,将反应液冷却至室温并加入0.1 mmol的六水合高氯酸亚铁(0.363 g),反应溶液立即变成紫红色,于室温下继续搅拌2 h。反应完全后,过滤除去杂质,滤液经乙醚扩散,得到配合物1和2。配合物fac-Δ-[Fe(S-L1)3][ClO4]2(1):深紫色晶体1,产率:82%。UVVis(CH3CN)λmax:218,287,545 nm。IR(KBr,ν/cm-1):3 116,2 986,2 935,1 575,1 496,1 451,1 384,1 098, 697,631。C45H48Cl5FeN9O8的元素分析计算值(%):C 50.23;H 4.50;N 11.72;实验值(%):C 50.40;H 4.41;N 11.59。配合物mer-Λ-[Fe(R-L2)3][ClO4]2·Et2O(2):红色晶体2,产率:77%。UV-Vis(CH3CN)λmax:206,290,545 nm。IR(KBr,ν/cm-1):3 187,2 986,2 923,1 623, 1 489,1 457,1 384,1 112,704,625。C50H62Cl2FeN9O8.50的元素分析计算值(%):C 57.09;H 5.94;N 11.98;实验值(%):C 57.01;H 5.86;N 11.89。
1.3 配合物的X-射线单晶衍射
配合物1和2的单晶衍射测试是在173(2)K温度下,使用BRUKER SMART APEXⅡDUO CCD单晶衍射仪测定。采用石墨单色化的Mo Kα射线(λ=0.071 073 nm),以φ-ω扫描方式收集数据。衍射数据用SAINT软件[21]还原,用SADABS软件进行吸收校正。晶体结构用SHELXS-97程序采用直接法解出并基于F2用SHELXL-97程序对所有非氢原子坐标及其各向异性参数进行了全矩阵最小二乘法精修[23]。
CCDC:1020959,1;1020960,2。
2 结果与讨论
2.1 晶体结构描述
为了直观说明手性席夫碱配合物的结构,我们测定了配合物1和2的X-射线单晶结构,晶体结构信息和键长键角见表1和2。X-射线单晶结构清晰地显示手性铁(Ⅱ)配合物1和2由于取代基R1与R2的不同形成了2种不同的结构。如图2a,2b所示,配合物1结晶于手性空间群P212121并形成fac-Δ构型的[Fe(S-L1)3]2+。每个结构基元中包含1个[Fe(L1)3]2+阳离子和2个高氯酸根阴离子。在每个化合物中,铁(Ⅱ)与3个不对称双齿手性席夫碱配体中的6个氮原子配位形成1个八面体配位环境。在173 K时配合物1的Fe-N平均键长为0.197 8 nm,表明铁(Ⅱ)在低自旋态,这与已报道的自旋转换化合物中描述的相一致[24-25]。在配合物1中,C6~C11、C36~C41、C21~C26组成3个苯环π平面,质心分别为Cg1、Cg3、Cg5。N7~N8与C31~C33、N4~N5与C16~C18、N1~N2与C1~C3组成3个咪唑环π平面,质心分别为Cg2、Cg4、Cg6。每个咪唑环与相邻配体上的苯环形成1个分子内π-π作用,质心间距的平均长度为0.375 5 nm。2个π平面的二面角的范围为5.135°~12.606°。虽然配合物1的结构中没有溶剂分子,但它仍然形成复杂而有趣的分子间相互作用。如图3a所示,在配合物1中,苯环上的C-H或C-Cl与相邻的苯环中心形成分子间C-H…π和C-Cl…π相互作用,C-H…π和C-Cl…π作用距离的平均长度分别为0.339 8 nm和0.330 0 nm。[Fe(L1)3]2+阳离子通过分子间C-H…π和C-Cl…π相互作用沿b轴形成1维超分子结构。在配合物2的Fe2中,C6ii~C11ii组成苯环π平面,质心为Cg1;N1ii~N2ii与C1ii~C3ii组成咪唑环π平面,质心为Cg1ii。同理,另外2个苯环π平面的质心为Cg2ii和 Cg3ii,另外2个咪唑环π平面的质心为H3ii和Cg3。配合物2的Fe1中,C6i~C11i组成苯环π平面,质心为Cg5i;N1i~N2i与C1i~C3i组成咪唑环π平面,质心为Cg5。同理,另外2个苯环π平面的质心为Cg4和Cg6,另外2个咪唑环π平面的质心为H3i和Cg6i。与1相比,配合物2形成了一个非常独特的结构。如图2c所示,配合物2结晶于手性空间群P43。每个结构基元中包含2个[Fe(L2)3]2+阳离子、4个高氯酸根阴离子和1个乙醚分子。在每个化合物中,铁(Ⅱ)与3个不对称双齿手性席夫碱配体中的6个氮原子配位形成一个八面体配位环境。咪唑基团在八面体的子午线上使配合物2形成mer构型。Fe(1)和Fe(2)与6个N原子形成的键长范围分别为0.205 7~0.220 0 nm和0.209 0~0.221 7 nm,表明配合物2在173 K时为高自旋态。每个[Fe(L2)3]2+基元中有2种不同类型的分子间作用力:咪唑环与相邻
配体上的苯环形成2个分子内π-π作用及咪唑环上的C-H与相邻配体上的苯环形成1个分子内CH…π作用。Fe(1)和Fe(2)的π-π作用质心间距的平均长度分别为0.364 1和0.366 5 nm,π平面的二面角的范围为3.962°~12.911°。C-H…π作用的平均长度分别为0.250 3和0.250 7 nm。配合物2的阳离子之间存在广泛的分子间相互作用形成了超分子网络结构。如图3b,每个[Fe(L2)3]2+阳离子与周围相邻的4个阳离子通过4个分子间C-H…π作用在ab平面上形成一个二维超分子网络结构。这4个分子间作用中2个来自于苯环与苯环的C-H…π作用(作用距离的长度分别为0.306 1 nm和0.324 6 nm),2个来自于苯环与咪唑环的C-H…π作用(作用距离的长度分别为0.297 5 nm和0.312 9 nm)。如果将每个[Fe(L2)3]2+阳离子看作1个4-连接节点,那么这个2D超分子结构可以看作是一个44网络拓扑结构(图3c)。在配合物1和2中,分子内的π-π堆积作用对其单一构型具有巩固作用,分子间的C-H…π和C-Cl…π作用稳固了整个晶体的手性堆积,从而使其结晶于手性空间群[26]。
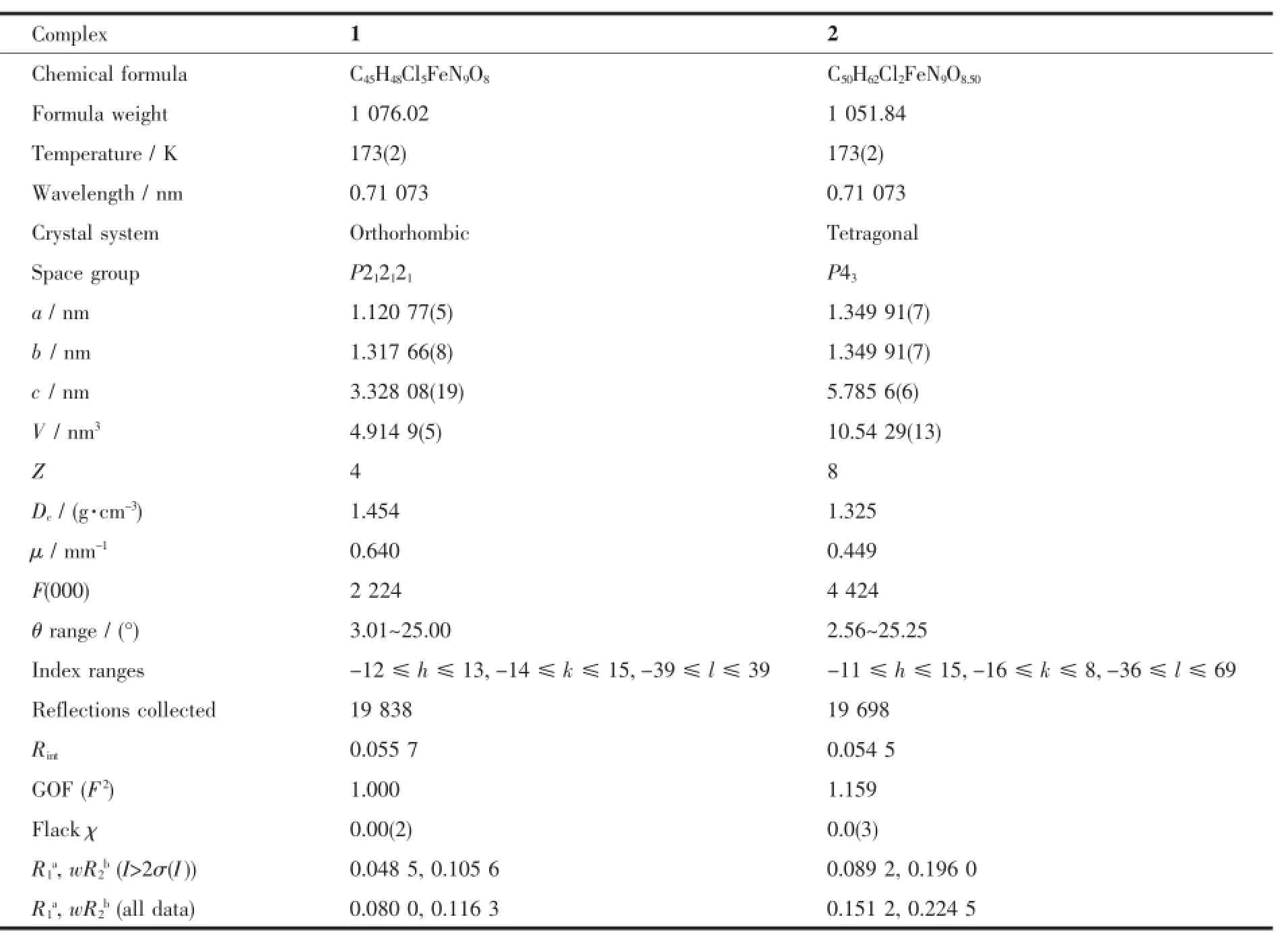
表1 配合物1和2的晶体学数据Table 1 Crystallographic data of complexes 1 and 2

表2 配合物1和2的主要键长(nm)和键角(°)Table 2 Main bond lengths(nm)and bond angles(°)data of 1 and 2
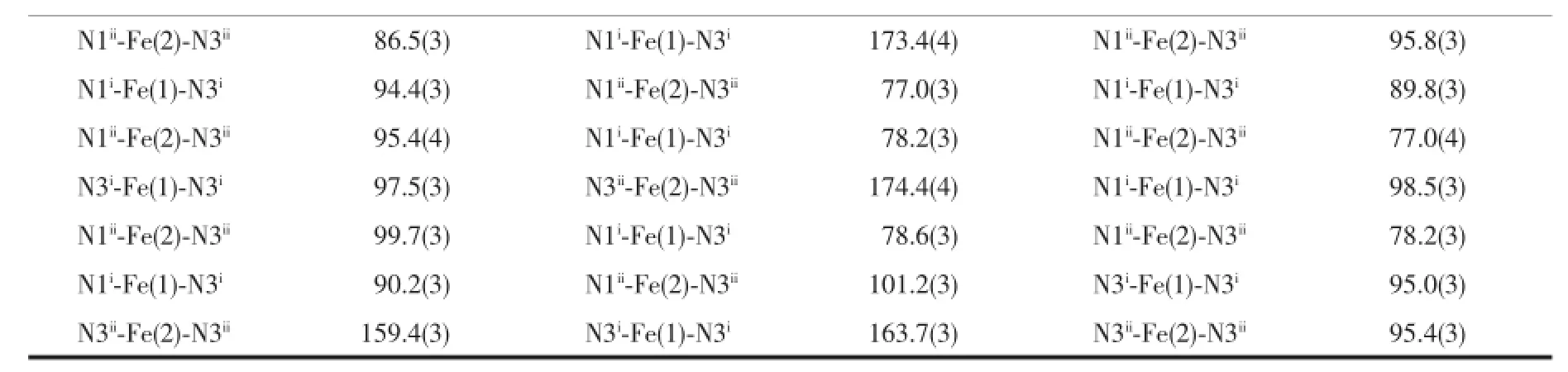
续表2
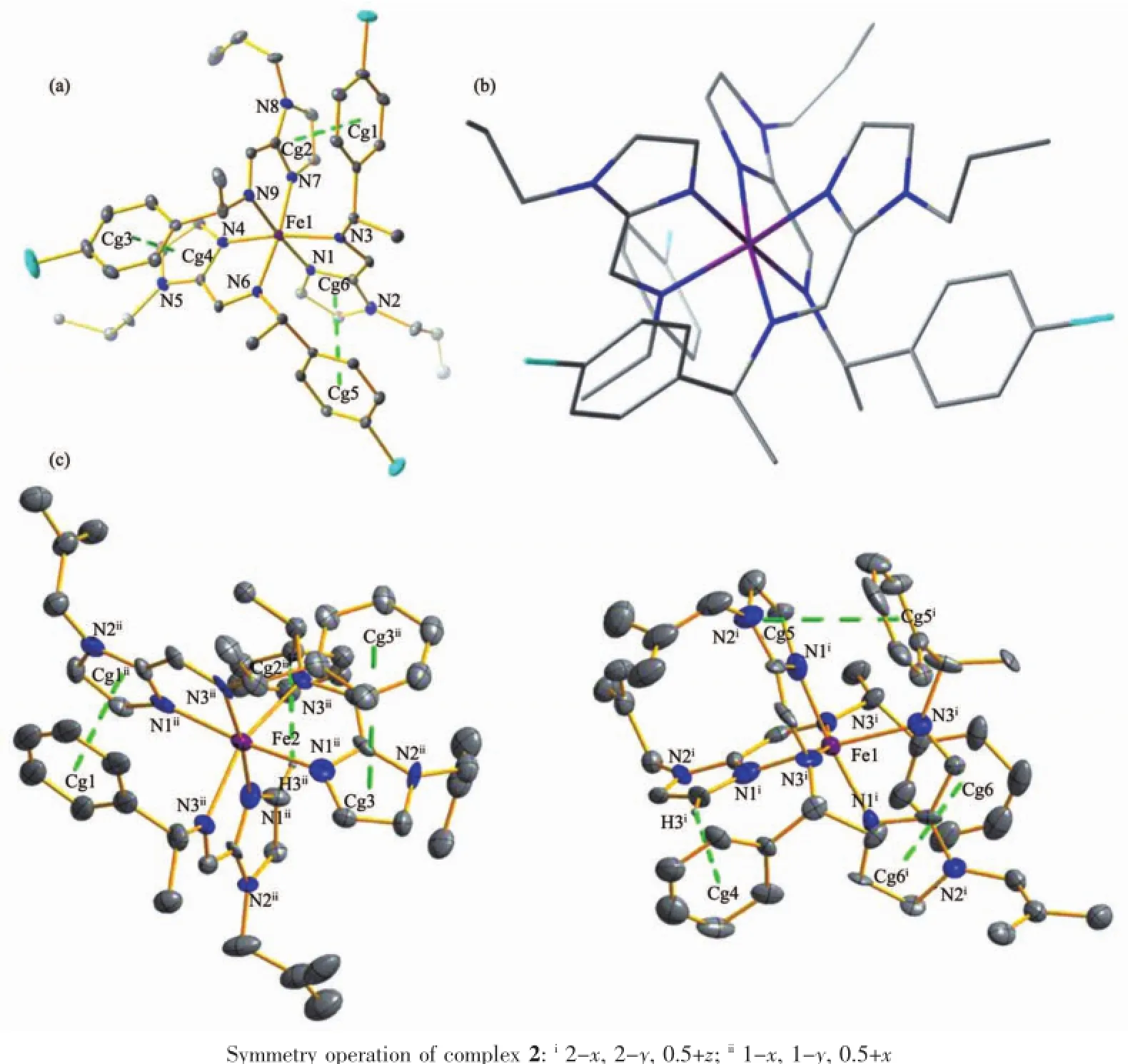
图2 配合物1(a)和2(c)的晶体结构椭球图,概率水平为30%;配合物1中fac-Δ-[Fe(L1)3]2+阳离子的侧面图(b)Fig.2 ORTEP diagrams showing the structures of complexes 1(a)and 2(c)with the atom numbering scheme at the30%probability levels;(b)Side view of the fac-Δ-[Fe(L1)3]2+cation in 1
2.2 光谱研究
配合物1,2的紫外光谱和圆二色谱在乙腈溶液中(浓度为10-5mol·L-1)测定,结果见图4。从图中可以看出,CD光谱有强烈的科顿效应,且其出峰位置与紫外光谱的出峰位置相一致。手性配体上的咪唑环和苯环的π-π*跃迁发生在低波数207~290 nm区域。光谱图中560 nm左右出现了d-d跃迁的金属配体电荷转移(MLCT)吸收峰,证明了配合物在溶液中的纯手性。与已报道的Λ和Δ构型[Fe(bpy)3]2+对映体的CD图谱比较得知,配合物1和2在溶液中是单一的Δ和Λ构型[27]。
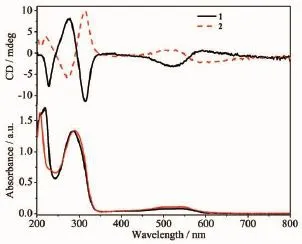
图4 配合物1,2在乙腈溶液中的CD(顶部)谱图和UV-Vis(底部)谱图Fig.4 CD(top)spectra and UV-Vis(button)of complexes 1 and 2 in CH3CN
2.3 磁性
利用MPMS-XL7 SQUID磁测量仪,在1 000 Oe的外加磁场下,以3 K·min-1的速率收集配合物1和2的多晶样品在2~400 K温度范围内升降温的磁性数据。直流磁化率结果如图5所示,配合物1和2因结构不同而表现出不同的磁性质。配合物1在高于298 K的高温区发生一步可逆自旋转换行为。降温时,磁化率的值从400 K时的3.42 cm3·K· mol-1迅速降低至300 K时的0.52 cm3·K·mol-1。表明配合物1在300~400 K温度区间发生自旋转换,自旋转换温度为372 K[28]。继续降温时,磁化率的值缓慢降低至2 K时的0.10 cm3·K·mol-1。与配合物1相比,配合物2表现出截然不同的一步自旋转换行为。2 K时,配合物2的磁化率值为0.18 cm3·K· mol-1。升温时,直到73 K磁化率的值几乎保持不变,然后迅速升高至232 K时的3.12 cm3·K·mol-1,接着缓慢升高至400 K时的3.39 cm3·K·mol-1。配合物2在73~232 K温度区间发生自旋转换,自旋转换温度为146 K。298 K时,配合物1和2的磁化率的值分别为3.24 cm3·K·mol-1和0.52 cm3·K·mol-1,表明在室温下配合物1中的铁(Ⅱ)金属中心处于低自旋状态,而配合物2中的铁(Ⅱ)金属中心处于高自旋状态[29]。众所周知,堆积方式和分子间作用对铁(Ⅱ)配合物的自旋转换行为有显著的影响,配合物1比2表现出更高的自旋转换温度归因于以上2个作用的协同影响[30-33]。配合物1比2结晶于对称性更低的手性空间群,且配合物1中存在强于C-H…π作用的C-Cl…π分子间作用力,导致配合物1的自旋转变向高温区移动。
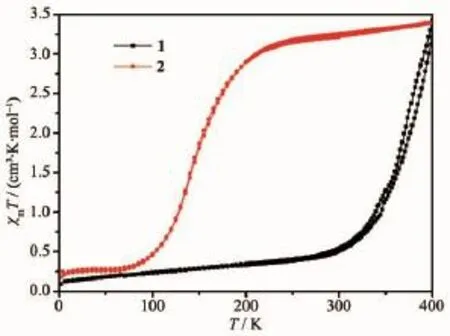
图5 配合物1和2的变温磁化率Fig.5 Plots ofχmT versus T for complexes 1 and 2
3 结论
在本文中,通过高氯酸亚铁,1-取代咪唑-2-甲醛和光学纯苯乙胺衍生物的多组分自组装成功合成了2个纯手性单核自旋转换铁(Ⅱ)化合物1和2。通过X-ray单晶衍射和圆二色谱探究其选择性的起源,证明在配合物1和2中,S型配体诱导[Fe(L1)3]2+阳离子形成单一fac-Δ构型,R型配体诱导[Fe(L2)3]2+阳离子形成单一mer-Λ构型。结构研究进一步表明[Fe(L1)3]2+阳离子形成特殊的分子内π…π相互作用,并通过分子间和C-Cl…π作用向空间延伸形成一维和二维超分子结构。[Fe(L2)3]2+阳离子形成特殊的分子内π…π和C-H…π相互作用,并通过分子间C-H…π作用向空间延伸形成二维超分子结构。磁性研究表明配合物1和2均表现出一步自旋转换行为,自旋转换的温度分别为372 K和146 K,证实了堆积方式和分子间作用对配合物的自旋转换行为有显著的影响。1和2的合成方法为纯手性自旋转换化合物的设计合成打开了新视角。
[1]Halcrow M A.Chem.Soc.Rev.,2011,40:4119-4142
[2]Gütlich P,Hauser A,Spiering H.Angew.Chem.,Int.Ed., 1994,33:2024-2054
[3]Guionneau P.Dalton Trans.,2014,43:382-393
[4]Rikken G L J A,Raupach E.Nature,1997,390:493-494 [5]Barron L D.Nature,2000,405:895-896
[6](a)Marcelo C,Sara G F,Ramon G M J.Adv.Mater.,2012,24: 3120-3123
(b)CaiH L,Zhang Y,Fu D W,etal.J.Am.Chem.Soc.,2012, 134:18487-18490
(c)Xu G C,Zhang W,Ma X M,et al.J.Am.Chem.Soc., 2011,133:14948-14951
[7]Train C,Gheorghe R,Krstic V,et al.Nat.Mater.,2008,7: 729-734
[8](a)Fu D W,Zhang W,Cai H L,et al.Angew.Chem.Int.Ed., 2011,50:11947-11951
(b)Liu C M,Xiong R G,Zhang D Q,etal.J.Am.Chem.Soc., 2010,132:4044-4045
(c)Cui H B,Wang Z M,Takahashi K,et al.J.Am.Chem. Soc.,2006,128:15074-15075
(d)Ohkoshi S I,Tokoro H,Matsuda T,et al.Angew.Chem. Int.Ed.,2007:3302-3305
[9]International Union of Pure and Applied Chemistry,Nomenclature of Inorganic Chemistry,Recommendation.Oxford: Blackwell Scientific Publications,1990.
[10]Cynthia P S,Suzanne E H,Guy J C,et al.Dalton Trans., 2010,39:4447-4454
[11]Alexander J.M,William C,Andrew S,et al.Dalton Trans., 2014,43:71-84
[12]Suzanne E H,Peter S.Dalton Trans.,2011,40:10268-10277 [13]Lacour J,Torche-Haldimann S,Jodry J J,et al.Chem. Commun.,1998:1733-1734
[14]Gao E Q,Yue Y F,Bai S Q,et al.J.Am.Chem.Soc.,2004, 126:1419-1429
[15]Tabellion F M,Seidel S R,Arif A M,et al.Angew.Chem., Int.Ed.,2001,40:1529-1532
[16]Rajesh C,Partha S M,Peter J S.J.Am.Chem.Soc.,2011, 111:6810-6918
[17]Wu J J,Cao M L,Bao H.J.Am.Chem.Soc.,2010,46:3687 -3689
[18]Suzanne E H,Laura E N,Nikola P C,et al.Dalton Trans., 2011,40:10416-10433
[19]Suzanne E H,Laura E N,Nikola P C,et al.Chem.Commun., 2009:1727-1729
[20]Suzanne E H,Guy J C,Alan D F,et al.Dalton Trans.,2013, 42:14967-14981
[21]SAINT-Plus,Version 6.02,Bruker Analytical X-ray System, Madison,WI,1999.
[22]Sheldrick G M.SADABS,An EmpiricalAbsorption Correction Program,Bruker Analytical X-ray Systems,Madison,WI, 1996.
[23]Sheldrick G M.SHELXTL-97,University of Göttingen, Göttingen,Germany,1997.
[24]Nishida Y,Kino K,Kida S.Dalton Trans.,1987,5:1157-1161
[25]Koenig E.Prog.Inorg.Chem.,1987,35:527-623
[26]Crassous J.Chem.Soc.Rev.,2009,38:830-845
[27]VankóG,Glatzel P,Pham V T,et al.Angew.Chem.Int. Ed.,2010,49:5910-5912
[28]Yamada M,Hagiwara H,Torigoe H.Eur.J.Inorg.Chem., 2006,12:4536-4549
[29]Nishi K,Matsumoto N,Iijima S.Inorg.Chem.,2011,50: 11303-11305
[30]Verdejo B,Gil-Ramirez G,Ballester P.J.Am.Chem.Soc., 2009,131:3178-3179
[31]Matouzenko G S,Jeanneau E,Verat A Y,et al.Dalton Trans.,2011,40:9608-9618
[32]Takahashi O,Kohno Y,Nishio M.Chem.Rev.,2010,110: 6049-6076
[33]Zhang W,Zhao F,Liu,T,et al.Inorg.Chem.,2007,46:2541-2555
Two Homochiral Spin-Crossover Iron(Ⅱ)Complexes Based on Bidentate Imidazole Schiff Base Ligands
GU Ling1REN Dong-Hong1LIU Zhi-Ming1SUN Xiao-Li1QIU Dan1GU Zhi-Guo*,1,2Li Zai-Jun1
(1School of Chemistry and Material Engineering,Jiangnan University,Wuxi,Jiangsu 214122,China)
(2The Key Laboratory of Food Colloids and Biotechnology,Wuxi,Jiangsu 214122,China)
Based on the bidentate schiff base ligands involving phenyl and imidazole groups,two homochiral mononuclear spin-crossover iron(Ⅱ)complexes,fac-Δ-[Fe(S-L1)3][ClO4]2(1),me r-Λ-[Fe(R-L2)3][ClO4]2·Et2O(2)(L1= 1-parachlorophenyl-N-(1-n-propylenyl-1H-imidazol-2-ylmethylene)ethanamine;L2=1-phenyl-N-(1-iso-butenyl-1H-imidazol-2-ylmethylene)ethanamine)have been synthesized.The two complexes have been determined by singlecrystal X-ray diffraction analysis,elemental analysis,IR spectra,1H NMR spectra,UV spectra and CD spectra. X-ray crystallography revealed thatthe iron(Ⅱ)center in 1 and 2 assumed an octahedralcoordination environment with 6 N donor atoms from three unsymmetrical bidentate chiral schiff base ligands.Each unit contained one [Fe(Ln)3]2+cation and two ClO4-anions in 1.While 2 contained two[Fe(Ln)3]2+cation,four ClO4-anions and one diethyl ether molecular.[Fe(Ln)3]2+components were chiral with single configuration due to the screw coordination arrangement of the chiral ligand around Fe(Ⅱ)centers.The Fe(Ⅱ)-N bond distances indicated that the Fe(Ⅱ)sites of 1 were in low-spin state,while the Fe(Ⅱ)centers of 2 were in high-spin state.As for[Fe(L n)3]2+,intramolecularπ-πinteractions were present between phenyl group and imidazole ring of an adjacent ligand.In 1 and 2, supramolecular architectures were formed through intermolecular C-H…πand/or C-Cl…πinteractions.Circular dichromism spectra confirmed the presence of non-racemic chiral metal centers in solution for complexes 1 and 2.Magnetic measurements revealed that 1 and 2 displayed obviously spin-crossover behaviour at 372 K and 146 K,respectively.Complexes 1 and 2 crystallized in differentpacking modes and intermolecularinteractions,therefore their SCO bahaviors were different.CCDC:1020959,1;1020960,2.
homochiral;spin-crossover;iron(Ⅱ)complexes;schiff base
O614.81+1
A
1001-4861(2015)07-1357-08
10.11862/CJIC.2015.183
2015-03-04。收修改稿日期:2015-05-08。
国家自然科学基金(No.21101078,21276105),新世纪优秀人才计划(No.NCET-11-0657),江苏省双创团队资助项目。*
