氘代阿戈美拉汀的合成
2015-11-27梁大伟王悦秋王艳艳
梁大伟,王悦秋,章 斌,王艳艳
(1.雅安职业技术学院 药学检验系,四川 雅安 625000;2.江苏联合职业技术学院 连云港中医药分院 中医药系,江苏 连云港 222007)
阿戈美拉汀,化学名称为N-[2-(7-甲氧基-1-萘基)乙基]乙酰胺,是法国Servier公司研发的第一个褪黑激素类抗抑郁药,于2009年2月19日经EMA(European Medicines Agency)批准于欧盟上市,用于治疗重度抑郁症,疗效明显,并能有效改善睡眠参数和保持性功能.药理研究表明阿戈美拉汀直接参与调节位于下丘脑的视交叉上核(SCN)的活动,激动褪黑激素MT1和MT2受体,同时拮抗位于额叶皮质的5-HT2C受体,校正昼夜节律,使节律重建,表现出抗抑郁作用[1].临床评价发现其对重度抑郁症(MDD)的疗效显著(25~50mg),但与安慰剂相比未见差异[2].
稳定同位素由于无放射性,物性相对稳定,将其标记到药物中,可运用气质联用(GC-MS)或液质联用(LC-MS)进行检测,具有较高的灵敏性[3].近年来,稳定同位素标记药物被广泛的应用于药代动力学的研究中,为了进一步开展对阿戈美拉汀的药代动力学研究,本文作者拟合成稳定同位素氘标记的阿戈美拉汀(1).
阿戈美拉汀的合成方法较多:1)以7-甲氧基-1-四氢萘酮为原料,由于已经存在基本母核,只需在其羰基位引入氨基侧链,如采用雷福尔马茨基反应策略[4-6];2)以2-(7-甲氧基萘-1-基)乙醇为原料,采用常规方法将分子中的羟基转变为氨基,然后酰化即可得到目标化合物[7];3)以7-甲氧基-1-萘酚为原料,先与三氟甲磺酸酐成酯,然后丙烯酰胺在醋酸铅催化下增长碳链并得到氨基取代中间体,最后经过还原、酰化得到目标化合物[8];4)以3-甲氧基苊醌为原料,在有机碱作用下开环得到乙基胺侧链,进一步将侧链中羰基还原、乙酰化得到目标化合物[9];5)以1-氨基-7-萘酚为原料,将氨基重氮化、碘代、罗森蒙德-冯布劳恩反应引入醛基取代,然后在碱性条件下与硝基甲烷缩合延长碳链,最后经过还原、乙酰化反应得到目标化合物[10];6)以β-萘酚为原料,通过傅克酰基化、维尔格罗特-金德勒反应引入8位侧链[11].鉴于上述合成路线存在的缺点与不足,以及氘代位置引入的需求,本文作者对氘代阿戈美拉汀的合成进行了改进.如图1所示,以苯酚为原料,经过氘代碘甲烷成醚、傅克酰基化、羰基还原、分子内傅克酰基化成环、克脑文盖尔缩合、2,3-二氯-5,6-二氰基-1,4-苯醌(DDQ)氧化芳构化、还原及氘代乙酰化反应得到目标化合物.该合成路线简洁、原料易得、反应条件易于控制.

图1 氘代阿戈美拉汀(1)的合成Fig.1 Synthesis of agomelatine-d6(1)
1 实验部分
1.1 仪器与试剂
Thomas-Hoover型熔点仪,温度计未加校正;核磁采用Bruker ARX-400型核磁共振仪测定,TMS为内标;质谱采用Agilent-6120GC/MS质谱仪测定,ESI源;高效液相色谱采用Agilent 1200 HPLC液相色谱仪测定(Zorbax Eclipse XDB-C18 column,5μm,4.6mm×250mm).氘代碘甲烷(CD3I)与氘代醋酸酐((CD3CO)2O)购于Sigma-Aldrich试剂公司,纯度与氘代丰度都大于99%,其他原料购于国药化学试剂有限公司,分析纯;常规溶剂为分析纯,未经过进一步处理,直接使用.
1.2 合成步骤
1.2.1 氘代苯甲醚-d3(2)的合成
将50%的氢化钠(7.2g,0.15mol)溶于80 mL的N,N-二甲基甲酰胺溶液中,室温下,向其中缓慢加入苯酚(14.1g,0.15mol),约30min加完,继续搅拌30min;然后向上述溶液中一次性加入氘代碘甲烷(12.1mL,0.19mol),反应液继续在室温下搅拌反应3h,最后倒入水中淬灭反应;乙酸乙酯萃取(3×20mL),合并有机相,依次用适量10%氢氧化钠溶液洗涤两次,水洗涤两次,饱和食盐水洗涤一次,无水硫酸镁干燥,减压蒸馏得到14.3g的氘代苯甲醚-d3(2),收率为85%,b.p.:153~155℃(与文献值[12]一致).
1.2.2 氘代4-(4-甲氧基)苯基-4-羰基丁酸-d3(3)的合成
将化合物2(12.2g,0.11mol)与丁二酸酐(15.0g,0.15mol)加入到250mL的反应瓶中,置于冰浴中搅拌,再分批加入三氯化铝(40.0g,0.30 mol),加毕,室温下搅拌反应1h,然后升温至60℃继续搅拌反应5h.反应完毕后,将反应瓶置于冰浴下,缓慢加入2%稀盐酸溶液200mL淬灭反应,析出大量浅绿色固体,抽滤.将滤饼溶于10%氢氧化钠溶液中,用适量石油醚洗涤三次.水溶液用5%稀盐酸调pH=2左右,即可析出大量白色固体,抽滤,干燥得24.7g白色粉末状固体氘代4-(4-甲氧基)苯基-4-羰基丁酸-d3(3),收率为73.7%,m.p.:146~147℃(文献值[13]:146℃).
1.2.3 氘代4-(4-甲氧基)苯基丁酸-d3(4)的合成
将化合物3(20.8g,0.10mol)、80%水合肼(16mL)与氢氧化钠(20.0g,0.5mol)溶于100 mL的一缩二乙二醇中,升温至110℃搅拌反应1 h,然后继续升温至175℃,常压蒸馏除水,待无水分蒸出时,反应完毕.将反应液降至室温,然后将反应液倒入适量碎冰中,用5%稀盐酸调水溶液的pH=2左右,析出大量固体,抽滤,干燥得18.0g白色固体粉末氘代4-(4-甲氧基)苯基丁酸-d3(4),收率为91.2%,m.p.:60~61℃(文献值[13]:60.5~61.5℃).
1.2.4 氘代7-甲氧基-1-四氢萘酮-d3(5)的合成
将五氧化二磷(228.0g,1.6mol)和磷酸(112 mL,85%)加入到1 000mL反应瓶中,升温至90℃搅拌反应5h,然后将溶于40mL CH2Cl2的化合物4(15.5g,0.08mol)滴加到上述反应瓶中,约30 min滴加完毕,并于80℃搅拌反应2h.反应完毕,向反应瓶中加入适量冰水淬灭反应液,用乙醚(3×30mL)萃取,合并醚层,依次用饱和碳酸氢钠和饱和食盐水洗涤,无水硫酸镁干燥,抽滤,减压浓缩得到12.5g淡黄色固体氘代7-甲氧基-1-四氢萘酮-d3(5),收率为92.3%,m.p.:61℃(文献值[13]:61℃),纯 度 为99.73%(HPLC),ESI-MS(m/z):180.1(M+H)+,1H NMR(400MHz,CDCl3)δ:2.13(2H,m,CH2),2.65(2H,t,J=6.0Hz,CH2),2.91(2H,t,J=6.0Hz,CH2),7.07(1H,dd,J=2.8,8.4Hz,Ar-H),7.15(1H,d,J=8.4Hz,Ar-H),7.50(1H,d,J=2.8 Hz,Ar-H).
1.2.5 氘代7-甲氧基-3,4-二氢-1-萘基乙腈-d3(6)的合成
将化合物5(8.8g,0.05mol)、氰基乙酸(6.4 g,75mmol)、苄胺(1.4g,13mmol)与庚酸(1.7 g,13mmol)溶于150mL的甲苯,回流分水约24 h,TLC检测至原料消失.反应完毕,冷却至室温,反应液依次用2mol/L的氢氧化钠溶液、水、饱和食盐水洗涤,无水硫酸镁干燥,抽滤,减压浓缩得9.4g油状氘代7-甲氧基-3,4-二氢-1-萘基乙腈-d3(6),冷冻过夜,固化,m.p.:48~49℃(文献值[6]为48~50℃),收率为94%.ESI-MS(m/z):203.1(M+H)+,1H NMR(400MHz,CDCl3)δ:2.31~2.36(2H,m,CH2),2.72(2H,t,J=8.0Hz,CH2),3.46(2H,d,J=1.6Hz,CH2),6.28~6.30(1H,m,Ar-H),6.66~6.67(1H,m,Ar-H),6.73~6.76(1H,m,Ar-H),7.08~7.10(1H,m,Ar-H).
1.2.6 7-甲氧基-1-萘基乙腈-d3(7)的合成
将2,3-二 氯-5,6-二 氰 基-1,4-苯 醌(DDQ)(7.6g,33.3mmol)溶于70mL的二氯甲烷,室温下向其中滴加溶于30mL二氯甲烷的化合物6(6.23g,30mmol),约30min滴加完毕,继续在室温下搅拌反应1h.反应完毕,过滤,紫黑色滤液依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,分出有机相,无水硫酸镁干燥,抽滤,减压浓缩得黄色固体粗品,乙醇-水(5∶3,体积比)重结晶得5.7g白色针晶体7-甲氧基-1-萘基乙腈-d3(7),收率为96%,m.p.:82~83℃(文献值[13]:83℃),纯度为99.97%(HPLC),ESI-MS(m/z):201.1(M+H)+,1H NMR(400MHz,CDCl3)δ:4.05(2H,s,CH2),7.06~7.07(1H,m,Ar-H),7.23~7.22(1H,m,Ar-H),7.32~7.36(1H,m,Ar-H),7.79~7.81(2H,m,Ar-H).
1.2.7 氘代阿戈美拉汀-d6(1)的合成
将四氢铝锂(1.1g,0.03mol)溶于50mL的无水四氢呋喃溶液,置于冰浴下,向其中滴加溶于20mL的无水四氢呋喃的三氯化铝(4.0g,0.03 mol),约20min滴加完毕,继续搅拌10min,得白色浑浊液;然后继续在冰浴条件下,向上述浑浊液中滴加溶于20mL无水四氢呋喃的化合物7(3.0g,15 mmol),约30min滴加完毕,室温下继续搅拌反应1 h.反应完毕,置于冰浴下,加入0.1mol/L的氢氧化钠溶液(2.1g),室温搅拌30min,抽滤,滤液用无水硫酸镁干燥,抽滤,减压浓缩得2.8g淡黄色油状粗品8,无需纯化,直接应用于下一步反应.
将上述淡黄色油状物8(2.8g,13.7mmol)溶于50mL干燥的二氯甲烷溶液,加入三乙胺(0.25 g,17.5mmol),冰浴下加入氘代醋酸酐(2.2g,20.4mmol),常温下继续搅拌反应约2h,TLC监测.待反应完毕后,反应液依次用饱和碳酸氢钠溶液和饱和食盐水洗涤,分出有机层,无水硫酸镁干燥,抽滤,减压浓缩,得白色固体粗品,进一步用乙醇-水(3∶5,体积比)重结晶,最终得3.1g针状结晶氘代阿戈美拉汀-d6(1),收率为94.2%,m.p.:108~109℃(文献值[6]:108℃),纯度为99.97%(HPLC),同位素丰度>98%,ESI-MS(m/z):250.1(M+H)+,1H NMR(400MHz,CDCl3)δ:3.22~3.28(2H,m,CH2),3.60~3.60(2H,m,CH2),7.13~7.15(1H,m,Ar-H),7.26~7.30(2H,m,Ar-H),7.47~4.49(1H,m,Ar-H),7.69~7.00(1H,m,Ar-H),7.73~7.79(1H,m,Ar-H);13C NMR(100MHz,CDCl3)δ:23.4,33.2,40.5,55.7,102.3,118.3,123.7,126.2,127.5,129.2,130.2,133.3,133.6,157.7,170.4.
2 结果与讨论
2.1 催化剂种类对合成化合物6的影响
如表1所示,采用苄胺与庚酸催化时,可以有很好的收率且杂质较少,且原料可以反应完全,并无需纯化,直接用于下一步反应.采用单一的吡啶、哌啶或醋酸铵催化,反应几乎不发生.此外,实验中也观察到这几个单一催化剂催化时反应没有水带出,但可回收原料.采用吡啶与醋酸铵或哌啶与醋酸铵混合催化反应,反应时间较长,收率很低,杂质较多,原料很难反应完全.
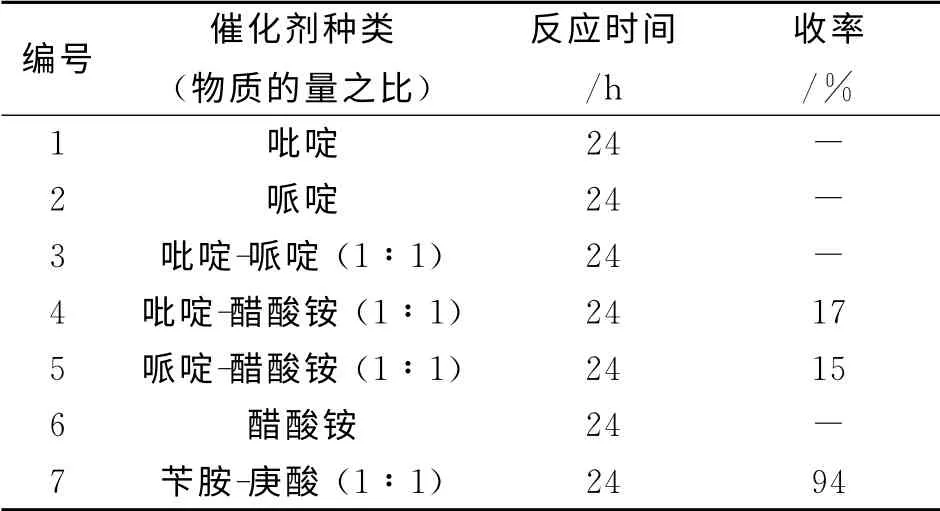
表1 催化剂种类对合成化合物6的影响Table 1 Effect of catalysts on the synthesis of compound 6
2.2 还原剂种类对化合物7中氰基还原氨化的影响
常用的氰基还原氨化方法有四氢铝锂和三氯化铝混合物还原[1]、氢化铝锂直接还原[14]、硼氢化钠在三氯化铝存在下还原[15]、硼氢化钠在六水合氯化钴的催化下还原[16].本文作者考察了多种催化剂对氰基还原氨化的还原效果.如表2所示,采用四氢铝锂与三氯化铝混合物的还原效果较好,收率较高,反应时间较短,得到的产物可以直接用于下一步的乙酰化,无需纯化.
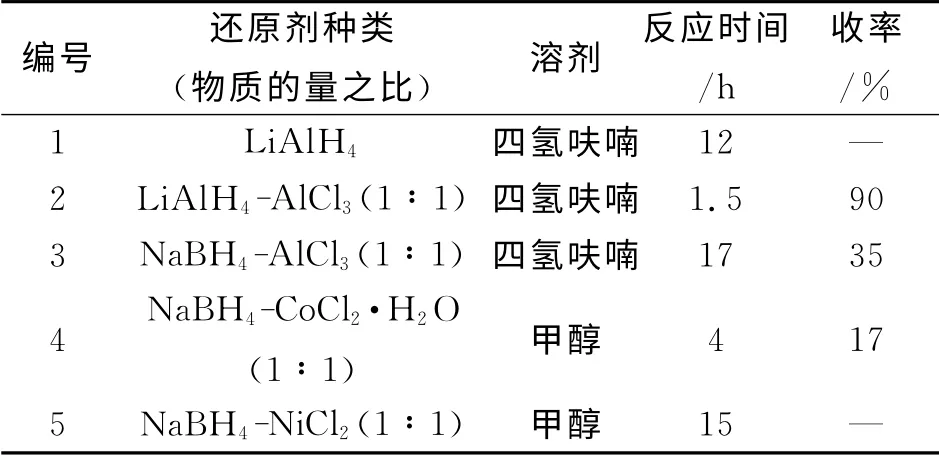
表2 还原剂种类对化合物7中氰基还原氨化的影响Table 2 Effect of reductants on the reduction of cyano group in compound 7
3 结论
以苯酚为起始原料,与氘代碘甲烷反应得到氘代甲基标记的苯甲醚,进一步经过傅克酰基化、克脑文盖尔缩合、芳构化、还原及氘代乙酰化反应得到最终目标化合物氘代标记的阿戈美拉汀-d6(1).该合成路线简洁,原料易得、反应条件易于控制,总收率为15.2%,化学纯度为99.97%,氘代丰度大于98%,可满足进一步对阿戈美拉汀的药代动力学研究.
[1]MÉSANGEAU C,PÉRÈS B,DESCAMPS-FRANÇOIS C,et al.Design,synthesis and pharmacological evaluation of novel naphthalenic derivatives as selective MT1melatoninergic ligands[J].Bioorg Med Chem,2010,18(10):3426-3436.
[2]赵绪韬.上市新药[J].世界临床药物,2011,32(60):381.
[3]李高.稳定同位素标记药物在临床药代动力学研究中的应用[J].中国临床药理学杂志,2000,16(1):58-62.
[4]JEAN A,RAYMOND H,SAID Y,et al.Naphthalene derivatives,procedure for their preparation and pharmaceutical compositions containing them:EP,0447285[P].1991-09-18.
[5]胡文浩,徐勤耀,杨琍苹.一种阿戈美拉汀的合成方法:中国,101792400[P].2010-08-04.
[6]JEAN-CLAUDE S,ISAAC G B,GILLES T,et al.Novel process for synthesizing and anovel crystal form of agomelatine as well as pharmaceutical preparations containing these:EP,1564202[P].2005-08-17.
[7]张桂森,陈道鹏,马彦琴,等.阿戈美拉汀的制备方法及其中间体:中国,101638376[P].2010-02-03.
[8]HARDOUIN C,LECOUVE J,BRAGNIER N,et al.Process for the synthesis of agomelatine:US,2010036162[P].2010-02-11.
[9]CHRISTOPHE H,JEAN P L.Process for the synthesis of agomelatine:US,2010036163[P].2010-02-11.
[10]KANDAGATLA B,RAJU V V,REDDY G M,et al.A facile synthesis of melatonergic antidepressant agomelatine[J].Tetrahedron Lett,2012,53(52):7125-7127.
[11]VUJJINIA S K,KRISHNAM RAJU D V R,BADARLA K R,et al.Total synthesis of agomelatine via Friedel-Crafts acylation followed by Willgerodt-Kindler reaction[J].Tetrahedron Lett,2014,55(29):3885-3887.
[12]KRUSE L I,DEBROSSE C W,KRUSE C H.Study of aromatic functional group conformations in solution by nuclear overhauser enhancement and relaxation techniques:detection ofπ-electron density and correlation with chemical reactivity[J].J Am Chem Soc,1985,107(19):5435-5442.
[13]CHANG H M,CHENG K P,CHOANG T F,et al.Structure elucidation and total synthesis of new tanshinones isolated from Salvia miltiorrhiza Bunge(Danshen)[J].J Org Chem,1990,55(11):3537-3543.
[14]CHU G H,LI P K.Synthesis of naphthalenic melatonin receptor ligands[J].Synth Commun,2011,31(4):621-629.
[15]俞建光,刘竺云,刘华权,等.一种阿戈美拉汀的制备方法:中国,102408350[P].2012-04-11.
[16]孟秀君,赵兴旺,刘丹,等.阿戈美拉汀的合成工艺改进[J].中国药物化学杂志,2011,21(2):138-140.
