抗肿瘤靶点PARP-1及其抑制剂的研究进展
2015-11-24谢怡悦石天晨王璐莹徐晓伟李阳辉林克江
谢怡悦,石天晨,王璐莹,徐晓伟,李阳辉,林克江
(中国药科大学药学院,江苏 南京 210009)
·前沿与进展·
抗肿瘤靶点PARP-1及其抑制剂的研究进展
谢怡悦,石天晨,王璐莹,徐晓伟,李阳辉,林克江*
(中国药科大学药学院,江苏 南京 210009)

聚二磷酸腺苷核糖聚合酶-1(PARP-1)参与DNA损伤修复,是近年来肿瘤治疗领域的热门靶点。从PARP-1的结构和作用机制出发,综述PARP-1抑制剂的主要结构类型及优化思路,展望其应用前景和亟需解决的问题。
PARP-1;抑制剂;肿瘤
在细胞的生长过程中,DNA不可避免地会受到外源或内源因素的作用产生DNA损伤。因此,细胞必须建立多种DNA损伤发现和修复体系,使受损的DNA得到及时的修复,以维持细胞的正常生理功能。
2015年诺贝尔化学奖获得者之一托马斯·林达尔提出的“碱基切除修复(base excision repair, BER)”机制已成为共识。被称为DNA修复酶的聚二磷酸腺苷核糖聚合酶 [poly(ADP-ribose) polymerase, PARP],密切参与DNA单链损伤的碱基切除修复,维持基因组稳定,近年来已成为肿瘤治疗领域的热门靶点。
PARP存在于细胞核内,是参与聚腺苷二磷酸核糖(poly(ADP-ribose), PAR)合成的酶,即一种ADP-核糖通过核糖基化-核糖键相连的多聚体。该酶由Chambon等人于1963年首次报道[1],但未引起重视。直至发现ADP-核糖基化参与蛋白的修饰才引起广泛的关注。
PARP包括18种亚型,它们具有很高的同源性,结构相似且能对许多核蛋白进行PAR修饰[2]。其中PARP-1在真核细胞内含量最高,对其结构和功能的研究也最为深入。
1 PARP-1的结构
PARP-1蛋白由长度为1 014个氨基酸的单条肽链构成(相对分子质量为 113 084),可划分为3个区域,分别为N端DNA结合域(DNA binding domain, DBD,2-372)、自身修饰域(automodification domain, AMD,373-524)和C端催化域(catalytic domain, CAT, 662-1 014)。DBD包含3个锌指结构域,其中ZnⅠ和ZnⅡ参与识别DNA损伤,ZnⅢ负责结构域间的联系,活化蛋白[3-4];AMD包含自身修饰的主要位点,其中有BRCA1(breast cancer type 1)的C端结构域 (385-476),参与结构域间的联系;CAT含有尼克酰二核苷酸(NAD+)的结合位点和合成PAR的催化位点,包括α螺旋结构域(helical subdomain, HD, 662-779)和二磷酸腺苷核糖转移酶结构域(ADP-ribosyl transferase, ART, 788-1 014)(见图1)。

图1 PARP-1的结构示意图Figure 1 Schematic structure of PARP-1
2 PARP-1的功能及作用机制
2.1 参与DNA损伤修复
当DNA损伤形成单链缺口(single strand break,SSB)时,PARP-1会直接参与BER过程。其作为感受器,DBD中的ZnⅠ和ZnⅡ结合到SSB上,进行DNA缺口的识别,并通过ZnⅢ活化形成同型二聚体。活化后的PARP-1催化NAD+降解为烟酸和ADP,再以ADP为底物在受体蛋白上合成PAR,调整其构象、稳定性和活性[5]。其中聚ADP核糖化的组蛋白和PARP-1自身带有大量负电荷,一方面与DNA链排斥分离,使染色质松弛,促进修复蛋白在DNA缺口处集结;另一方面又可招募并活化相关修复蛋白,如X线修复交叉互补组合-l(XRCC1)、DNA连接酶Ⅲ和DNA聚合酶β[2]。另外,PARP-1还可能与募集的蛋白中特异性PARP结合模序非共价结合,促进DNA损伤修复。
若SSB未及时修复,DNA会通过复制过程介导双链DNA断裂产生双链DNA缺口(double strand break,DSB)。在其下行通路中,p53蛋白作为决定细胞进一步修复或走向凋亡的分子开关,可能通过Sirtuin蛋白1(sirtuin type 1, SIRT1)的去乙酰化作用调节PARP-1活性[6]。在活化状态下,PARP-1可通过在蛋白上合成PAR来修饰ATM、MRE11等同源重组(homologous recombination, HR)修复蛋白,DNA-PKcs、Ku70/80等非同源末端连接(non-homologous end joining, NHEJ)修复蛋白和拓扑异构酶Ⅰ[2],促进HR修复和DNA-PK、Ku70/80、XRCC4、DNA连接酶等介导的NHEJ修复[5],并控制细胞周期、维持基因组完整性;反之,在抑制状态下,p53蛋白可激活细胞周期素依赖性激酶(cyclindependent kinase, CDK)和p21、Bcl-2等凋亡相关蛋白,分别促进细胞周期阻滞和Caspase依赖的细胞凋亡(见图2)。
当DNA损伤过于严重时,PARP-1过度活化,在自身聚ADP核糖基化的过程中,耗竭细胞能量(NAD+、ATP),使凋亡诱导因子(AIF)向核移动,介导非Caspase依赖的细胞凋亡[7]。另外,PARP-1还参与促进核苷酸切除修复(nucleotide excision repair, NER)[8]。
2.2 作为细胞凋亡信号
在Caspase依赖的细胞凋亡早期,被激活的半胱天冬酶将PARP-1切割出相对分子质量分别为24 000和89 000的2个片段。这在许多类型的细胞中被认为是细胞凋亡的信号[3]。可能的机制是:包含DBD的相对分子质量为24 000的片段与DNA缺口结合,阻碍修复蛋白向染色体募集;包含AMD和CAT的相对分子质量为89 000的片段无法被DNA损伤活化,阻碍DNA损伤信号传递,促进细胞凋亡[7]。
2.3 参与基因转录调控
PARP-1在多条通路中通过对组蛋白进行PAR修饰,使染色质松弛,促进基因转录;或通过与转录因子相互作用,调控基因转录。如PARP-1可作为NF-κB的转录辅激活因子,激活NF-κB相关基因的转录[9]。
3 PARP-1抑制剂的作用机制
PARP-1抑制剂针对PARP-1参与SSB/DSB修复的功能,通过抑制PARP-1催化活性和将PARP-1捕获于损伤的DNA上这2种机制发挥作用。一方面,PARP-1抑制剂通过与PARP-1的CAT竞争性结合,抑制其催化活性,使SSB得不到及时修复,产生DSB;另一方面,PARP-1抑制剂通过抑制PARP-1的自身PAR修饰、与CAT结合导致PARP-1变构,增强PARP-1与损伤DNA的结合强度,将PARP-1“捕获”于损伤DNA上(见图3[10]),使细胞核中其他PARP-1难以与损伤DNA结合,进一步阻断DSB的可能修复途径,促进细胞凋亡。有研究表明,PARP-1抑制剂捕获PARP-1的能力与其抑制肿瘤细胞的活性正相关[10-11]。
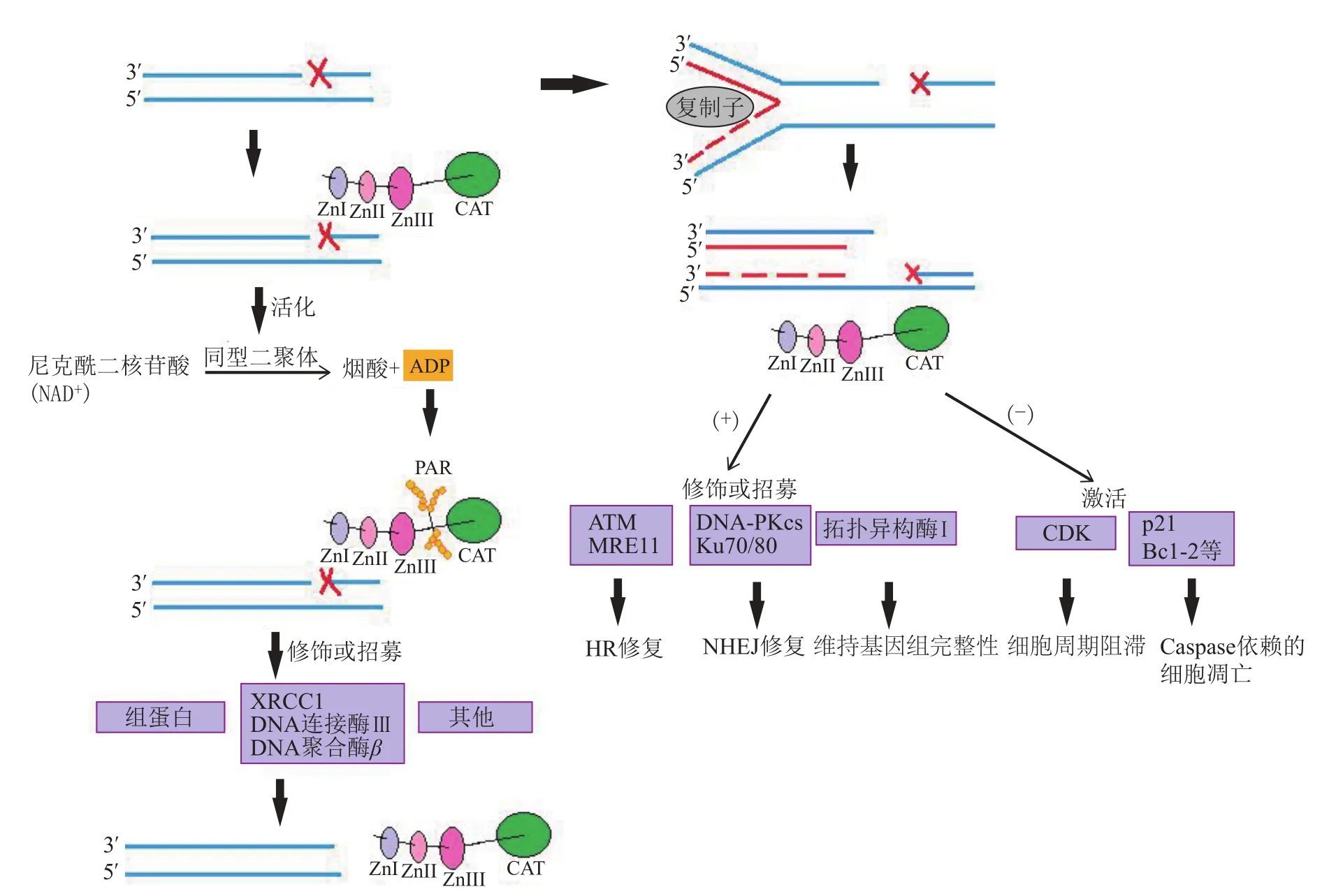
图2 PARP-1参与SSB/DSB修复的作用机制Figure 2 Mechanism of PARP-1 in SSB/DSB repair
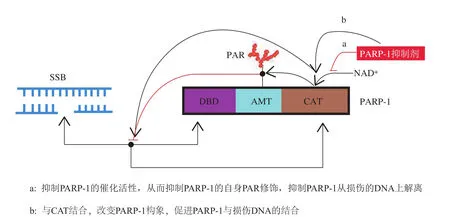
图3 PARP-1抑制剂于损伤DNA上捕获PARP-1的分子机制Figure 3 Molecular mechanism of PARP-1 inhibitors in trapping PARP-DNA complexes
近年来的研究发现,PARP-1抑制剂单药对BRCA1/2突变的乳腺癌及卵巢癌细胞有明显抑制作用[12-13]。根据图2所示机制,若肿瘤细胞存在HR修复缺陷(如BRCA1/2突变),DSB将无法修复,即导致所谓的PARP-1抑制剂和HR修复缺陷对肿瘤细胞的合成致死作用。HR修复是个复杂的过程,许多基因和蛋白成分参与其中,包括ATM、ATR、CHK1、EMSY、PTEN、RAD51及其同系物如FANC蛋白、MRE11、RAD50、NBS1等,BRCA1/2只是其中的重要成分之一。HR修复途径中的任一基因突变或表达沉默,即会引起HR修复途径缺陷,PARP-1抑制剂即可能通过合成致死作用发挥抗肿瘤活性。
另外,PARP-1抑制剂还可作为放(化)疗增敏剂发挥抗肿瘤作用。许多化疗药物(如烷化剂、铂类、拓朴异构酶Ⅰ/Ⅱ抑制剂等)和放疗均通过直接或间接损伤DNA来发挥对肿瘤细胞的杀伤作用。由于PARP-1在DNA损伤修复中起关键作用,可将PARP-1抑制剂作为放(化)疗增敏剂与放(化)疗联用,增强抗肿瘤疗效[14]。同时,还可因此减少放(化)疗用药或放射剂量,降低毒副作用。
4 PARP-1抑制剂的主要结构类型与优化策略
上世纪70年代,PARP-1抑制剂的研究就已开始,主要针对肿瘤、卒中、心脏缺血、炎症和糖尿病等疾病。目前研究的PARP-1抑制剂主要针对PARP-1的SSB修复功能,应用于肿瘤治疗,自2003年以来已有多个候选药物进入临床研究阶段。许多公司在第1代PARP-1抑制剂(指各公司第1批启动临床前及临床研究的PARP-1抑制剂)的基础上,从选择性、PARP-1捕获能力、抗药性等方面进行优化,设计并合成了第2代PARP-1抑制剂(指各公司继第1代抑制剂后新投入临床前及临床研究的PARP-1抑制剂),目前也有部分进入临床研究[8]。
由于PARP-1的内源性底物为NAD+,目前进入临床的PARP-1抑制剂大多模拟NAD+中烟酰胺的结构,与CAT中的烟酰胺结合位点(NI)或腺苷(AD)位点结合[8],竞争性抑制PARP-1活性。虽然PARP-1抑制剂的结构各异,可分为多类,但是它们的先导化合物种类有限,主要分为以下3类。
4.1 取代苯甲酰胺
上世纪70年代,研究发现烟酰胺(NA,1)有轻微的PARP抑制作用(IC50=210 μmol·L-1)(Clark J B等,Biochim Biophys Acta, 1971年),但也作用于细胞内许多其他酶。于是人们以烟酰胺为先导化合物合成了一系列衍生物,包括苯甲酰胺(Shall S, Biochem J, 1975年)(2)及3-氨基苯甲酰胺(3-AB,3)、3-氨基苯甲酸(4)、3-氨基苯乙酮(5)、3-氨基-N-甲基苯甲酰胺(6)、2-氨基苯甲酰胺(7)、4-氨基苯甲酰胺(Purnell M R等,Biochem J, 1980年)(8)等,在50 μmol·L-1下对猪PARP的抑制率分别为96%、90%、10%、31%、0%、0%、0%,确证了芳酰胺基团和取代基位于酰胺间位对选择性抑制PARP的必要性。因此,苯甲酰胺和间位取代苯甲酰胺被作为模板进行进一步优化。

4.2 多环内酰胺
上世纪90年代初,Parke-Davis公司的一个研究团队以苯甲酰胺和间位取代苯甲酰胺为模板,通过比较5位和7位取代的二氢异喹啉酮活性来确定其优化构型,从而引入了二环内酰胺结构(Suto M J等,Anticancer Drug Des, 1991年)。实验结果确证了引入环结构来固定酰胺构象及5位引入杂原子取代的必要性(见图4)。随后,Banasik和Ueda领导的团队用牛PARP筛选了100多个二环及三环内酰胺化合物(Banasik M等,J Biol Chem, 1992年),发现了一些多环内酰胺骨架(9a~ 9d, 10a ~ 10c, 11 ~ 16, IC50分别为1.5、0.41、0.10、0.42、8.0、9.5、120、0.39、7、0.30、5.6、8.1、12 μmol·L-1),除骨架10外,二氢异喹啉酮(9)、异喹啉酮(11,12)、菲啶酮(13)、喹唑啉酮(14)、喹唑啉二酮(15)和2,3-杂氮萘酮(16)等日后均用于优化。其中,2,3-杂氮萘酮是最重要的骨架,经优化合成了3个进入临床研究的PARP-1抑制剂[15]。
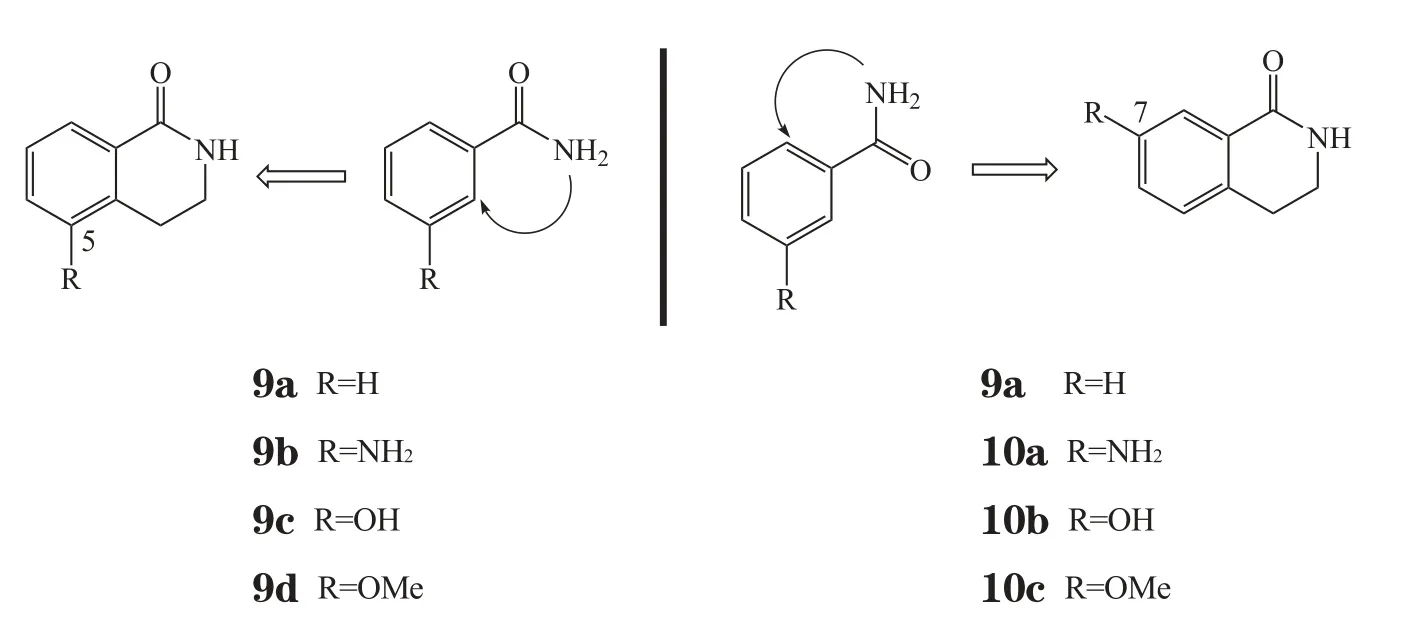
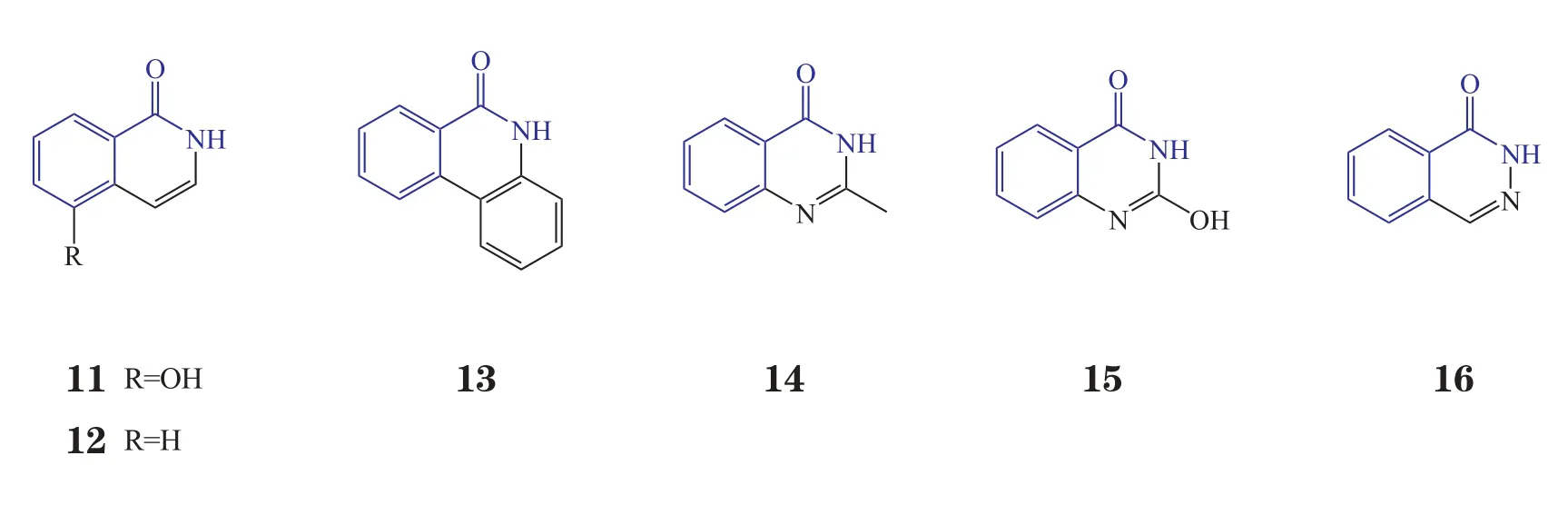
图4 二环内酰胺结构及优势取代位置的发现Figure 4 Discovery of bicyclic aryl amides and the optimal orientation of the amide with respect to substituents on the aryl ring
随着上世纪90年代中期多环内酰胺骨架的发现,PARP-1抑制剂的构效关系初步建立。数年后,一些第1代PARP-1抑制剂与鸡PARP-1的复合晶体结构被测定(Ruf A等,Biochemistry, 1998年),PARP-1抑制剂的构效关系得到进一步完善:1)与PARP-1催化活性位点上Ser904和Gly863结合的部位具备合适的氢键受体和供体(如酰胺中的羰基和N端活泼H),成环或“假环”可提高结合能力;2)具备可与PARP-1中Tyr896和 Tyr907形成π-π相互作用的富电子芳香环或杂环平面结构,故芳酰胺更适宜;3)A环上酰胺间位有能与Glu988形成氢键的氢键供体和受体(如杂原子取代基);4)A环上酰胺临近位置处有Ala898和Lys903组成的小疏水口袋,可有小疏水基团取代(如 CH3、 F、 Cl等);5)抑制剂分子右下部分应设计大疏水基团,与PARP-1上NAD+结合部位的大疏水口袋(AD位点)结合,以提高抑制剂活性、水溶性和其他理化性质(见图5)。
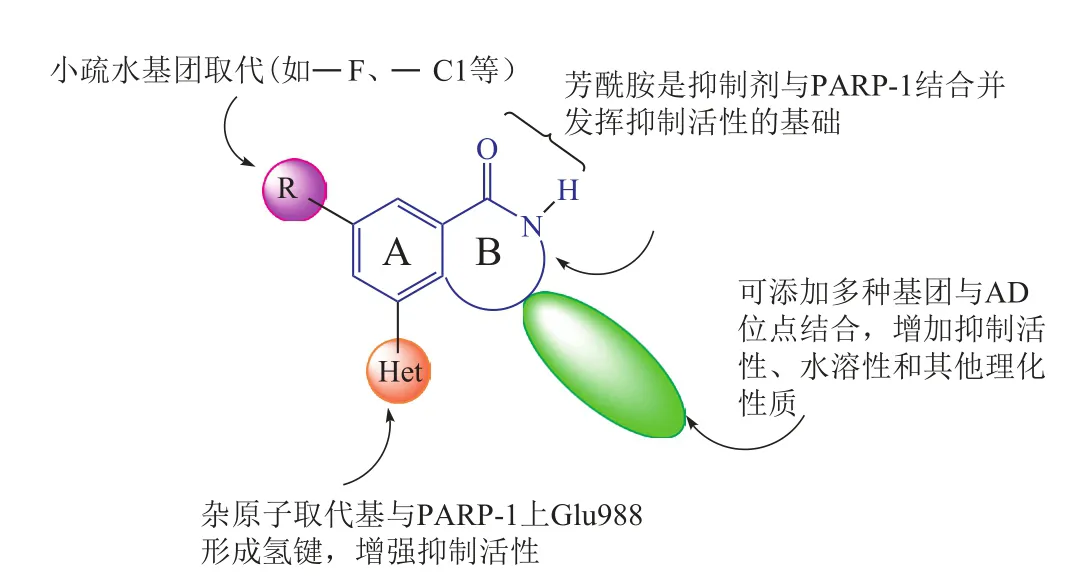
图5 PARP-1抑制剂的构效关系Figure 5 SAR of PARP-1 inhibitors
4.3 苯并咪唑甲酰胺
上世纪90年代中期,Golding和Griffin领导的团队设计并合成了苯并咪唑-4-甲酰胺(见图6)这一由分子内氢键构成的“假环”结构骨架,既保证了酰胺间位的杂原子取代基(图6中金色部分),又限制了酰胺的构象,如化合物17a和17b(IC50分别为1.1和0.1 μmol·L-1)(Griffin R J等, Anticancer Drug Des, 1995年)。这一重要骨架后经优化合成了3个PARP-1抑制剂且均已进入临床研究。
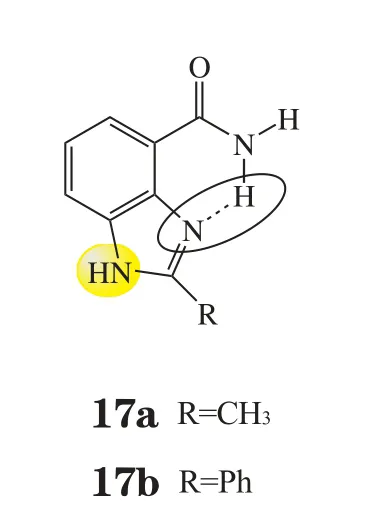
图6 苯并咪唑甲酰胺骨架Figure 6 Benzimidazole carboxamides as potent PARP inhibitor scaffolds
5 PARP-1抑制剂的研究现状
现有的以上述骨架为先导化合物,根据已知构效关系合成的许多第1代抑制剂在选择性、耐药性等方面存在问题,而第2代抑制剂开始研发时间不长,故目前尚无第2代PARP-1抑制剂成功上市。目前,处于临床研究阶段的具有抗肿瘤疗效的PARP-1抑制剂及有望进入临床的活性化合物举例如下。
5.1 奥拉帕尼和第2代PARP抑制剂AZD2461
21世纪初,Maybridge和Kudos公司合作,在化合物16的4位引入苄基,得到苗头化合物4-苄基-2,3-杂氮萘酮(18,IC50=770 nmol·L-1)[16],不仅使活性增强了10倍,而且合成方便。为进一步提高抑制剂酶水平上的活性(IC50<10 nmol·L-1)和细胞活性(如透膜性),提高水溶性和口服生物利用度,须对化合物18进一步优化。在侧链苯环3位引入丁二酰亚胺提高了酶水平上的活性和细胞活性[17]、增强了代谢稳定性;4位氟取代显著提高了化合物的透膜性(PF50=5.6),得到先导化合物19(KU58684,IC50=5 nmol·L-1)。再将3位的丁二酰亚胺替换成二酰基取代的哌嗪基团,使化合物的水溶性和口服生物利用度显著提高,得到临床候选药物奥拉帕尼(olaparib,KU59436,AZD2281,20,IC50=5 nmol·L-1,PF50=25.8)。奥拉帕尼与甲基磺酸甲酯(MMS)、替莫唑胺(TMZ)联用可抑制肿瘤组织的增殖,在BRCA缺陷的癌细胞系中单独使用也有明显活性[16]。2013年,阿斯利康(AstraZeneca)公司对奥拉帕尼展开了3项Ⅲ期临床研究,分别研究其用于治疗BRCA突变的转移性乳腺癌患者、BRCA突变的卵巢癌患者和铂敏感复发性卵巢癌患者的疗效。2014年5月,该公司以在铂敏感复发性卵巢癌患者组中进行的随机Ⅱ期临床研究(19号研究)结果为主要依据,向美国FDA递交了奥拉帕尼的新药申请,并获得优先审评资格。但由于相关专家质疑19号研究过程中招募BRCA基因突变患者的群体过小,所提供数据无法充分说明其药效,且考虑到其潜在不良反应,暂缓批准奥拉帕尼的上市,要求公司提供进一步详细说明数据。2014年12月19日,FDA通过加速审批通道批准了奥拉帕尼胶囊(商品名:Lynparza)上市(EMA于2014年12月16日批准),准其作为单药治疗之前至少经过3次化疗的BRCA基因突变的晚期卵巢癌妇女,并伴随批准一款名为BRACAnalysis CDx的基因检测试剂,用于检测卵巢癌患者的血样中是否存在BRCA基因突变。目前,奥拉帕尼联合紫杉醇治疗胃癌患者的临床研究已进入Ⅲ期,针对前列腺癌和肺癌患者的临床研究已进入Ⅱ期。阿斯利康公司仍在准备公布卵巢癌Ⅲ期临床研究的最新研究数据,以获得FDA的完全批准。
由于在临床研究中观察到患者对奥拉帕尼易产生抗药性,阿斯利康公司推出了与其结构相似的第2代抑制剂AZD2461(21)。该药与奥拉帕尼性质相似,但在抗药性、耐受性方面有明显改善,且可减少复发[18]。不过,2014年完成的Ⅰ期临床研究宣告失败。奥拉帕尼和AZD2461的优化过程见图7。
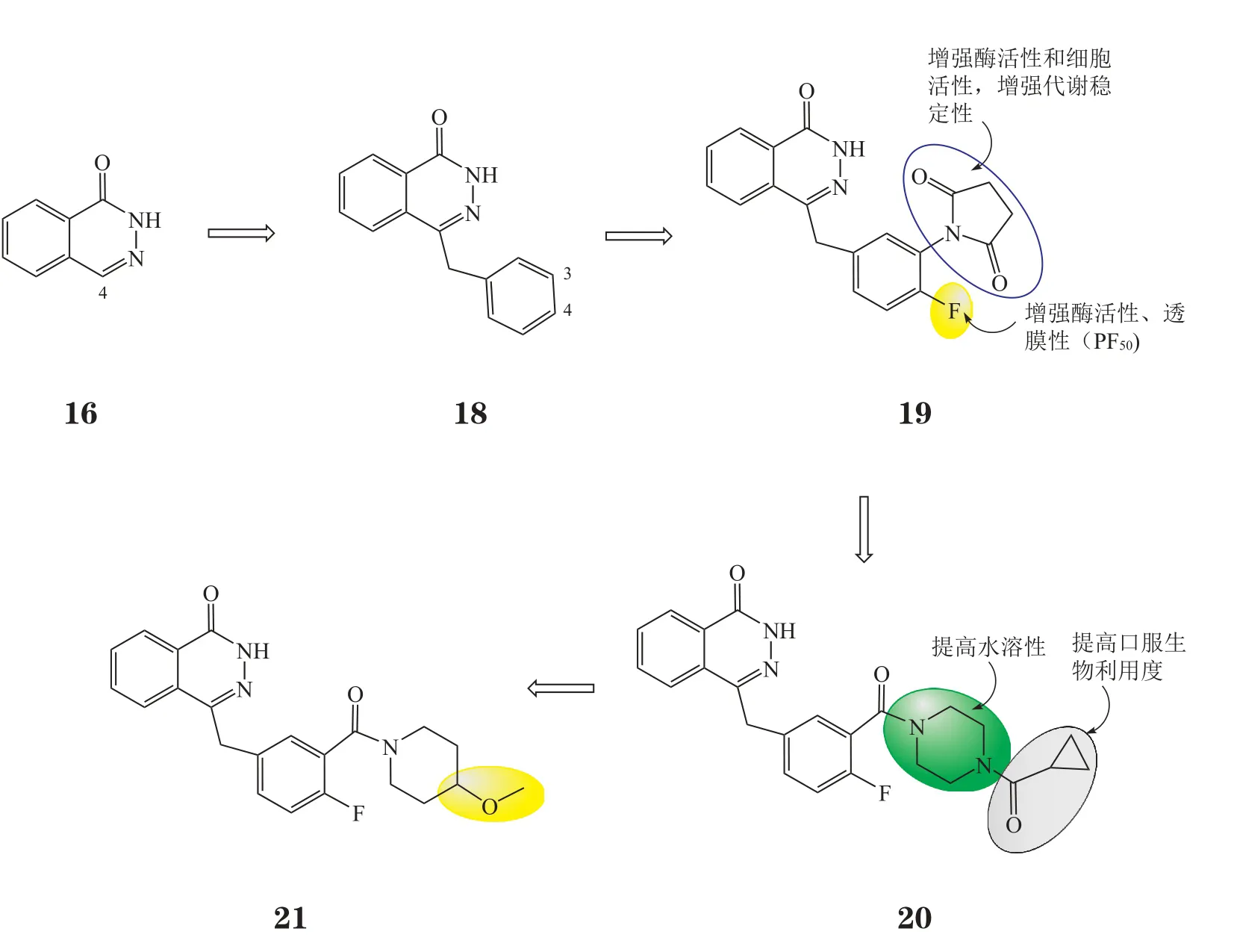
图7 奥拉帕尼和AZD2461的优化过程Figure 7 Evolution of olaparib and AZD2461
5.2 瑞卡帕尼
瑞卡帕尼是在苯并咪唑-4-甲酰胺(17c)的基础上以共价键形成内酰胺的结构,属于三环吲哚内酰胺类,是首个进入临床的PARP-1抑制剂。其优化过程基于PARP-1与抑制剂的复合晶体结构:先在2位引入芳环得到化合物22(NU1085,Ki=6 nmol·L-1),其与PARP-1中Tyr896和Tyr907形成π-π相互作用,体外活性显著增强,但水溶性较差。为提高水溶性和结构新颖性,在化合物22的芳环4位引入叔胺基团,并将酰胺基团用七元环固定成内酰胺结构,所得化合物23(AG014344,Ki=5.6 nmol·L-1,PF50=7.8)的水溶性、抑制活性和透膜性均有所提高[19]。在此基础上进一步优化得瑞卡帕尼(24,rucaparib,AG014699,PF-0137338,CO-338),酶水平上的活性(PARP-1,Ki=1.4 nmol·L-1)和细胞活性(PF50=8.1)均有所提高,其优化过程见图8。瑞卡帕尼与TMZ联用有很好的抗肿瘤活性,与其他活性相近的先导化合物相比具有显著优越性,于2003年作为化疗增敏剂率先进入临床研究阶段。但由于观察到较严重的骨髓抑制作用,与TMZ联用的Ⅲ期临床研究仍未启动。另外,瑞卡帕尼对PARP-1的选择性较差[20],甚至可作用于Akt /蛋白激酶B和Stat3等其他抗肿瘤靶点[21]。然而,分子分型技术的发展为瑞卡帕尼的研究提供了新的思路[22]。于2014年完成的一个Ⅱ期临床研究找出了瑞卡帕尼响应患者的生物标记物。从中选择几个特定的分子分型进行Ⅲ期临床研究,找出卵巢癌、输卵管癌和原发性腹膜癌患者中对瑞卡帕尼产生响应的特定基因型[23]。2015年4月,FDA授予瑞卡帕尼突破性治疗药物资格,使其可用于一些使用一线、二线药物疗法失败的BRCA基因突变的卵巢癌患者。瑞卡帕尼是PARP抑制剂中首个获此资格的药物。同时,瑞卡帕尼与顺铂联用治疗BRCA突变的三阴性乳腺癌的研究正处于Ⅱ期临床阶段。
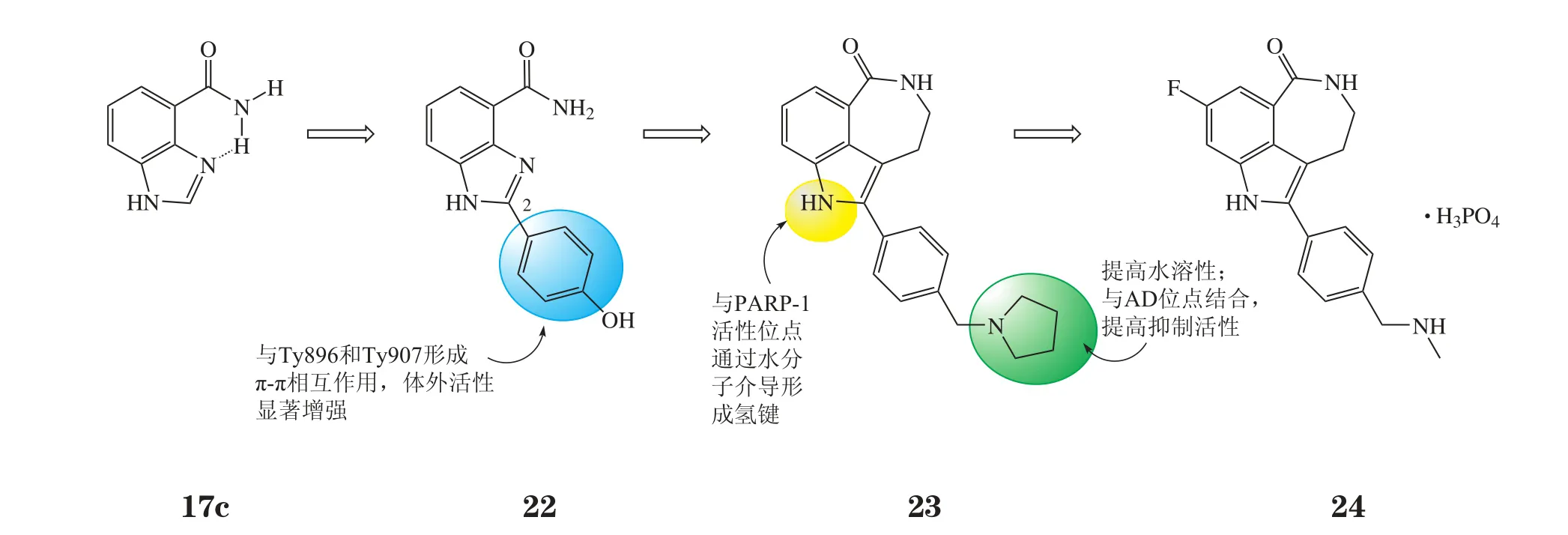
图8 瑞卡帕尼的优化过程Figure 8 Evolution of rucaparib
5.3 维利帕尼
维利帕尼也是在苯并咪唑-4-甲酰胺(17c)的基础上优化而来。2001年,雅培(Abbott)公司收购BASF公司,基于其研究基础,合成了数百个2位烷基胺取代的化合物[24-25],以对PARP-1和细胞(过氧化物损伤的C41细胞)的IC50低于10 nmol·L-1为条件进行筛选,选出临床前候选药物A-620223(25,Ki=8 nmol·L-1,EC50= 3 nmol·L-1)。其中叔胺基团保证了足够的水溶性(大于5 g·L-1)、哌啶N上的烷基侧链取代则显著增强了细胞活性。在构效关系研究过程中,雅培公司发现2位取代的碳原子若为季碳原子,则化合物的细胞活性将比叔碳原子取代的化合物高2~13倍[25]。于是,对A-620223进一步优化,得到临床候选药物维利帕尼(26,veliparib,ABT-888,Ki=5 nmol·L-1,EC50=2 nmol·L-1),其优化过程见图9。虽然维利帕尼(R构型)与其对映异构体(S构型)在酶水平上的活性相同(Ki=5 nmol·L-1),但由于R构型在不同物种中的口服生物利用度好于S构型(56%~92% vs 39% ~ 66%),故选用R构型。其中侧链二级胺的存在使维利帕尼能够透过血脑屏障,可用于脑瘤的治疗。由于可以显著增强TMZ和卡铂的抗肿瘤活性[25],维利帕尼于2006年进入临床试验阶段。目前,维利帕尼在与TMZ联用治疗脑瘤的临床研究中采用Ⅰ+Ⅱ期联合设计,与全脑放疗联用治疗转移性脑瘤已进入Ⅰ期临床,与卡铂联用治疗三阴性乳腺癌已进入Ⅲ期临床研究。
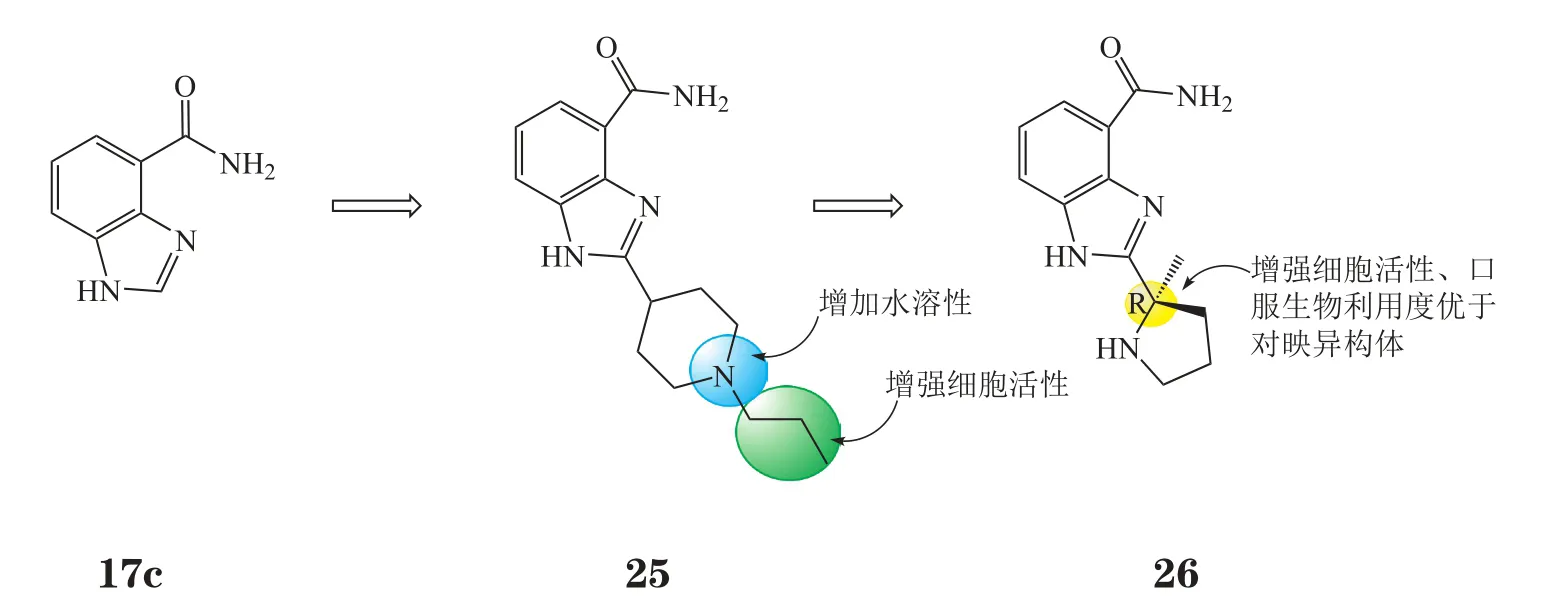
图9 维利帕尼的优化过程Figure 9 Evolution of veliparib
5.4 尼拉帕尼
尼拉帕尼是由默克(Merck)公司研发的临床候选PARP-1抑制剂(2012年后由Tesaro公司接手)。其优化过程同样以苯并咪唑-4-甲酰胺(17c)为起点,将咪唑环用多种五元氮杂环替换,分别测定在酶水平上的活性和细胞(过氧化物损伤的HeLa细胞)活性,最终选出药动学性质、在酶水平上的活性和细胞活性最佳的吲唑作为骨架[26]。其中,2位苯基取代的化合物27(IC50=24 nmol·L-1,EC50=3.7 μmol·L-1)活性较强,在大鼠和人微粒体内显示出较强的稳定性,在大鼠体内的口服生物利用度(41%)和半衰期(5.1 h)均较好,被选为先导化合物。为增强水溶性,在苯基对位引入仲胺基团,并以化合物在BRCA1敲除的HeLa细胞中的致半数细胞毒性的药物浓度(CC50)和在野生型HeLa细胞中的CC50的比值为参数,筛选出具有最佳细胞活性和BRCA选择性的化合物。临床候选药物尼拉帕尼(28,niraparib,MK-4827,IC50=3.8 nmol·L-1,EC50=4 nmol·L-1其优化过程见图10)在大鼠体内的口服生物利用度极佳(65%)、半衰期较长(3.4 h),与其对映异构体相比,BRCA选择性更好(CC50比值28 vs 11)。且尼拉帕尼除竞争性抑制PARP-1的催化活性中心外,其在DNA上捕获PARP-1的能力是临床候选药物中最强的[10],与维利帕尼等药物相比具有优势。2008年,尼拉帕尼进入临床研究,在复发性种系BRCA突变的卵巢癌和散发性非BRCA缺失的卵巢癌中进行了Ⅰ期临床研究,每日口服300 mg耐受性良好,主要不良反应为1/2级的贫血、疲劳和恶心[27-28]。目前,Tesaro公司分别在HER2缺陷的非种系BRCA缺陷乳腺癌患者和铂敏感的卵巢癌患者中开展尼拉帕尼单药维持治疗的Ⅲ期临床研究。
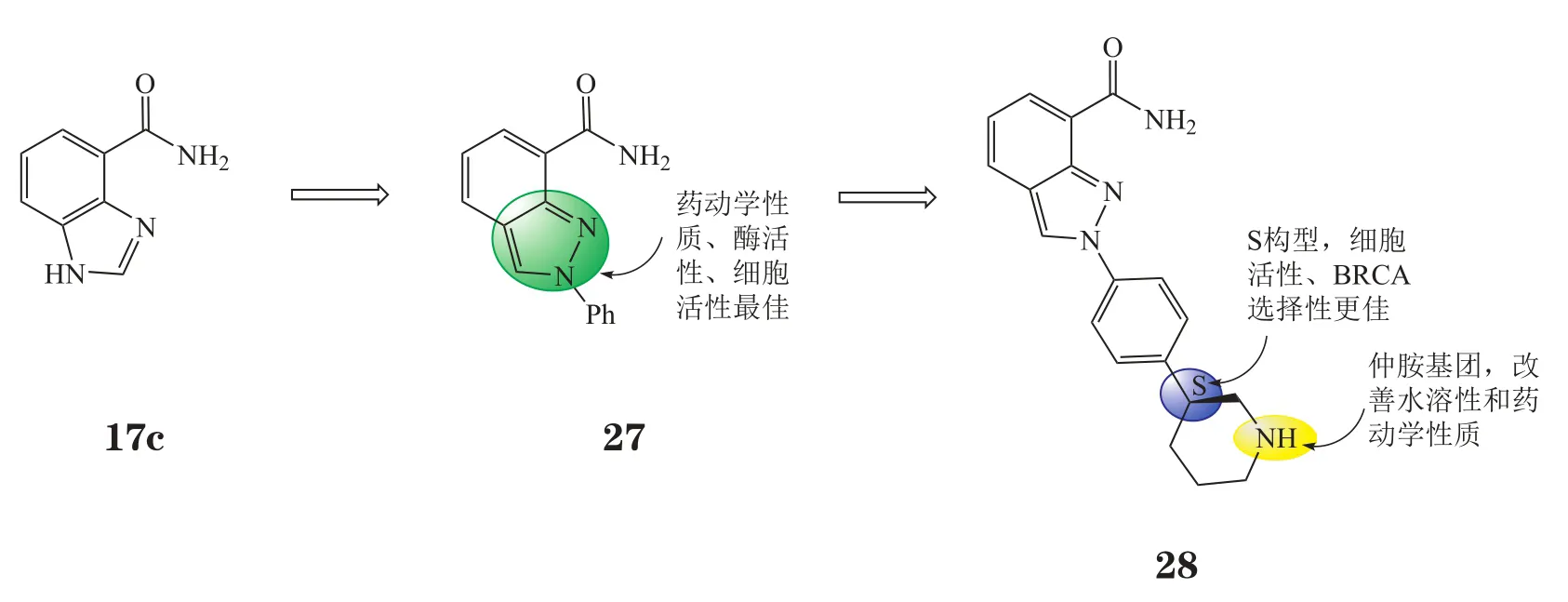
图10 尼拉帕尼的优化过程Figure 10 Evolution of niraparib
5.5 BMN-673
BMN-673是较晚进入临床的一种口服PARP-1抑制剂,其优化过程非常有趣。最初,Lead公司融合2,3-杂氮萘酮(16)和苯并咪唑-4-甲酰胺(17c)2种骨架,设计出一新的骨架——吡咯并二氮杂萘酮(29,见图11)。在设计合成路线时,以化合物30为起始原料,预想在碱性条件下第1步可得化合物31,加入肼后经3步反应即可得目标产物。但实际合成过程中第1步却得到化合物32,加入肼后得化合物33(见图12)。实验表明,此偶然发现的化合物33有很强的PARP-1抑制活性(IC50=6.1 nmol·L-1),于是Biomarin公司便以化合物33为先导化合物进行优化(见图13)。将一个取代苯环用咪唑环代替,得到新的先导化合物34(IC50=2.3 nmol·L-1,EC50=16.9 nmol·L-1),其酶水平上的活性和细胞(过氧化物损伤的LoVo细胞)活性均优于化合物33,且由于咪唑环的引入,水溶性更好。但与TMZ联用和单独作用于BRCA2缺陷型细胞株(Capan-1细胞)时,化合物34的活性并不尽如人意(IC50分别为89 和2 μmol·L-1)。于是,对化合物34进一步优化,在其结构的图示位置(金色圈出部分)增加2个氟原子和1个氮原子,得到临床候选药物BMN-673(35)。该化合物在酶水平上的活性(IC50=0.57 nmol·L-1)和细胞活性(EC50=2.51 nmol·L-1)与化合物34相比均有显著提高,对Capan-1细胞也有很强的抑制活性(IC50=5 nmol·L-1),且各项抑制活性均明显优于其对映异构体(36,BMN-674)。作为晚进入临床的第1代PARP-1抑制剂,BMN-673在酶水平上的活性是其他临床在研抑制剂的3 ~ 8倍,细胞活性可达20 ~ 200倍[11]。另外,它捕获PARP-1的能力很强,细胞毒性却并无显著提高[10],Ⅰ期临床推荐剂量为每天口服1 mg,是其他PARP抑制剂的几百分之一。它不仅可作为化疗增敏剂与TMZ和铂类药物联用,且可单药抑制BRCA缺陷的乳腺癌和卵巢癌细胞[29]。目前,BMN-673用于BRCA缺陷的转移性乳腺癌患者的试验正处于Ⅲ期临床阶段。
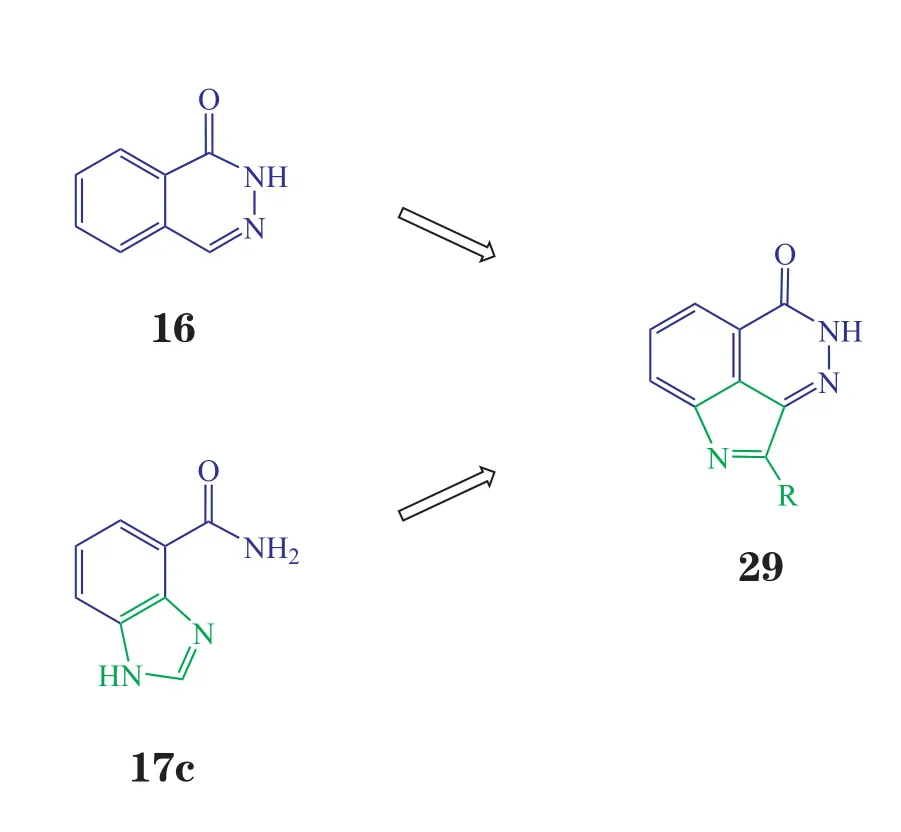
图11 吡咯并二氮杂萘酮骨架的初步设计Figure 11 Preliminary design of pyrrolophthalazinones
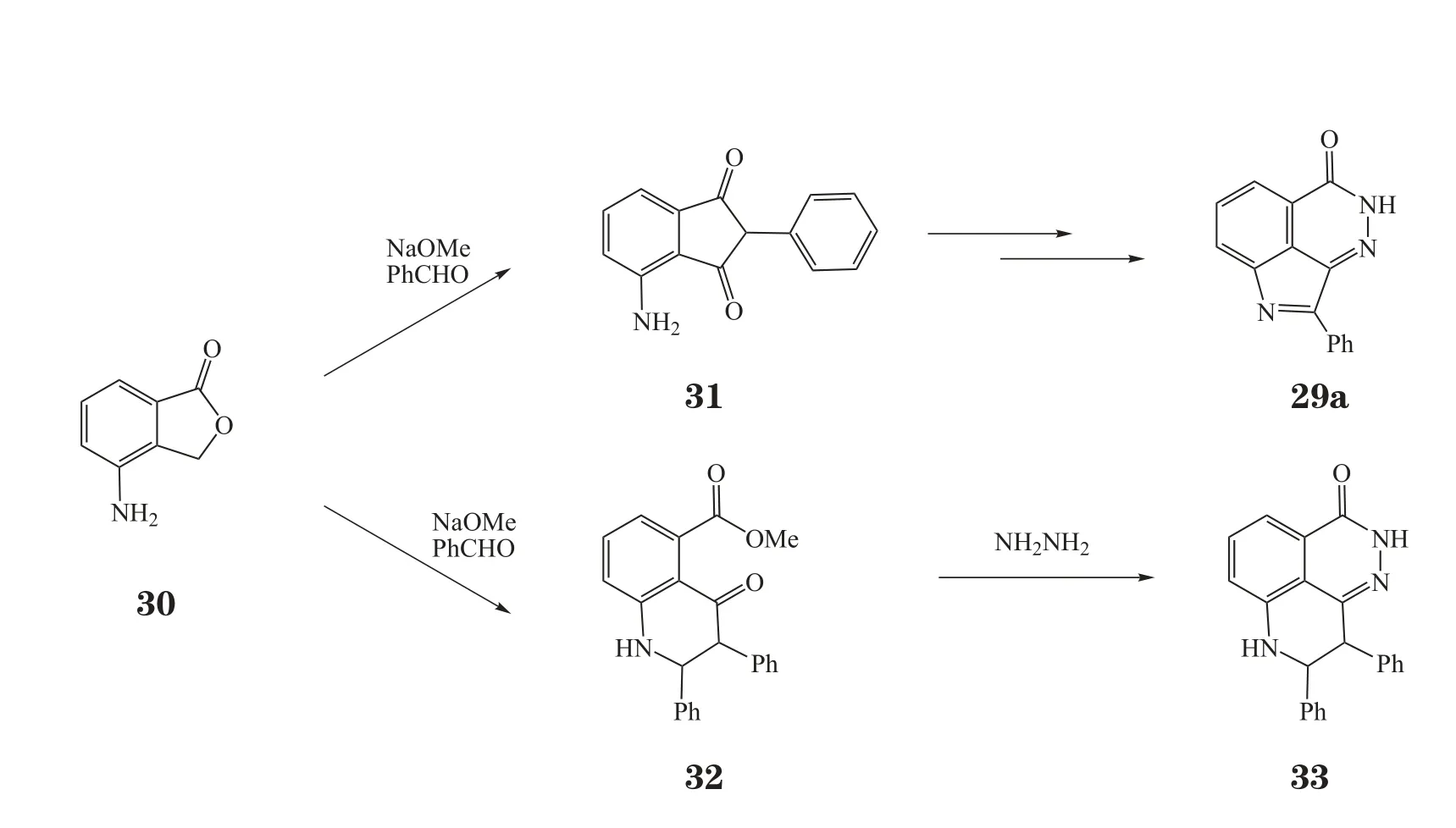
图12 先导化合物33的偶然发现Figure 12 Serendipity of compound 33
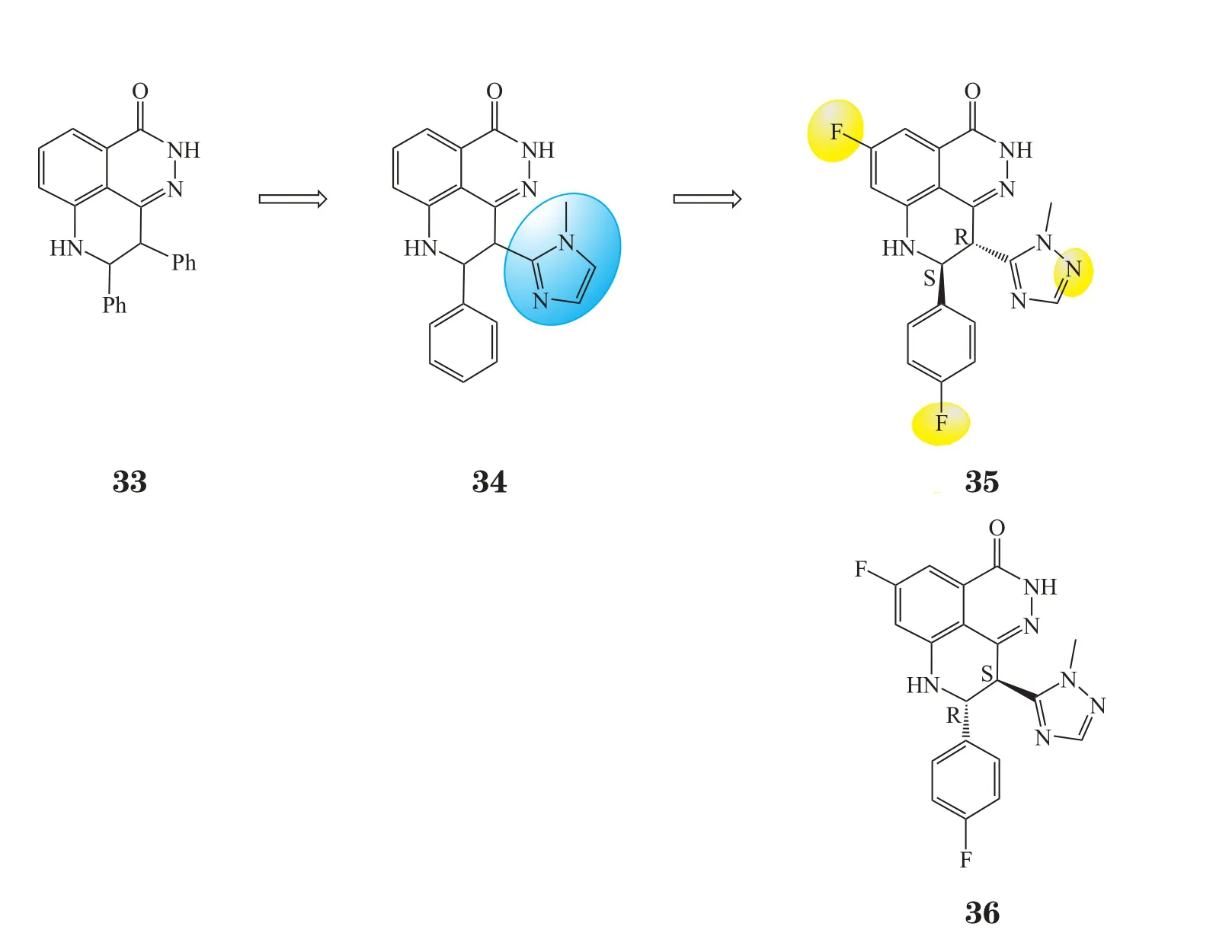
图13 BMN-673的优化过程Figure 13 Evolution of BMN-673
5.6 E7016和第2代PARP抑制剂E7449
2005年,Guilford/MGI的一个小组以2,3-杂氮萘酮(16)和四环异喹啉酮(37,GPI 6150,IC50=60 nmol·L-1)2种骨架为基础[30],设计出早期先导化合物38(GPI 15427,IC50=31 nmol·L-1),其苯环上的哌嗪取代改善了分子的水溶性和透膜性。将哌嗪基团替换成羟基哌啶,即得到临床候选药物E7016(39,GPI 21016,Ki=50 nmol·L-1),其优化过程见图14。在白血病鼠模型中,E7016与顺铂联用显示出显著的化疗增敏效果,联用组(E7016,40 mg·kg-1,ip)较顺铂组的鼠平均寿命延长了160%。在恶性胶质瘤异种移植模型中,E7016与TMZ合并放疗联用,显示出放(化)疗增敏活性。联用组(E7016,40 mg·kg-1,po,X射线4 J·kg-1,TMZ 3 mg·kg-1)与TMZ合并放疗的对照组相比,平均寿命延长了32%。Ⅱ期临床研究于2012年展开,研究E7016与TMZ联用治疗黑色素瘤的疗效。另外,第2代PARP抑制剂E7449(结构未公开)的Ⅰ期临床研究也已展开。
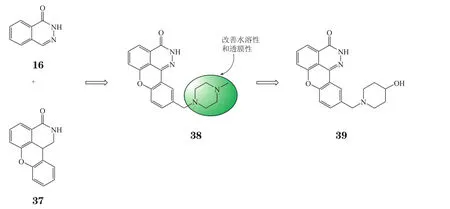
图14 E7016的优化过程Figure 14 Evolution of E7016
5.7 1位长链取代的苯并[1,7]八氢萘啶类化合物
2013年3月,Ye等[31]在对抗帕金森病药阿扑吗啡(40)的儿茶酚部分进行电子等排优化的过程中,偶然发现将儿茶酚替换成内酰胺后得到的化合物完全失去了多巴胺受体的激动活性。结合Torrisi等的报道,Ye等设计并合成了3个系列的1位长链取代的苯并[1,7]八氢萘啶类化合物(见图15),长链分为连接基团和末端效应基团两部分。第2系列以苯甲酰基为连接基团,其中使用2,3-杂氮萘酮作为末端效应基团的化合物41,表现出极强的PARP-1抑制活性(IC50=0.31 nmol·L-1),但在细胞内效力温和(BRCA2缺陷型细胞:CC50=96.0 nmol·L-1;BRCA1缺陷型细胞:CC50=23.3 nmol·L-1)。以该化合物为先导进行优化,得到化合物42(IC50=3.46 nmol·L-1,其优化过程见图16),其不但对PARP-1酶有效,而且在BRCA缺陷型细胞中有很好的效力(BRCA2缺陷型细胞:CC50=4.53 nmol·L-1;BRCA1缺陷型细胞:CC50=0.26 nmol·L-1)。
这一新型PARP-1抑制剂损伤了BRCA缺陷型细胞中的细胞周期进程,同时增强了TMZ等烷化剂类抗肿瘤药物的细胞毒性,具有成为临床候选药物的光明前景。
5.8 NMS-P118
2015年8月,Papeo等[32]报道了一种高选择性、可口服的PARP-1抑制剂NMS-P118。由于目前的临床候选PARP-1抑制剂多为PARP-1/2非选择型,意大利纳维亚诺医学研究院(NMS)在其化合物库中进行了高通量筛选,以发现高选择性的新型PARP-1抑制剂。研究人员将高通量筛选得到的化合物43进行优化,以酶水平上的活性(PARP-1 KD ≤0.1 μmol·L-1)和选择性(PARP-2 KD / PARP-1 KD约为100)为条件进行筛选,得先导化合物44(PARP-1 KD = 0.050 μmol·L-1,PARP-2 KD/PARP-1 KD >200)。为提高PARP-1酶活性,对化合物44进一步优化得NMS-P118(45,PARP-1 KD = 8 nmol·L-1,PARP-2 KD/PARP-1 KD > 83),其优化过程见图17。
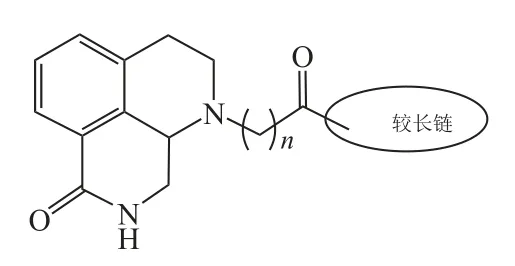
图15 1位长链取代的苯并[1,7]八氢萘啶类化合物Figure 15 Benzo[de][1,7]naphthyridin-7(8H)-ones bearing a functionalized long chain appendage
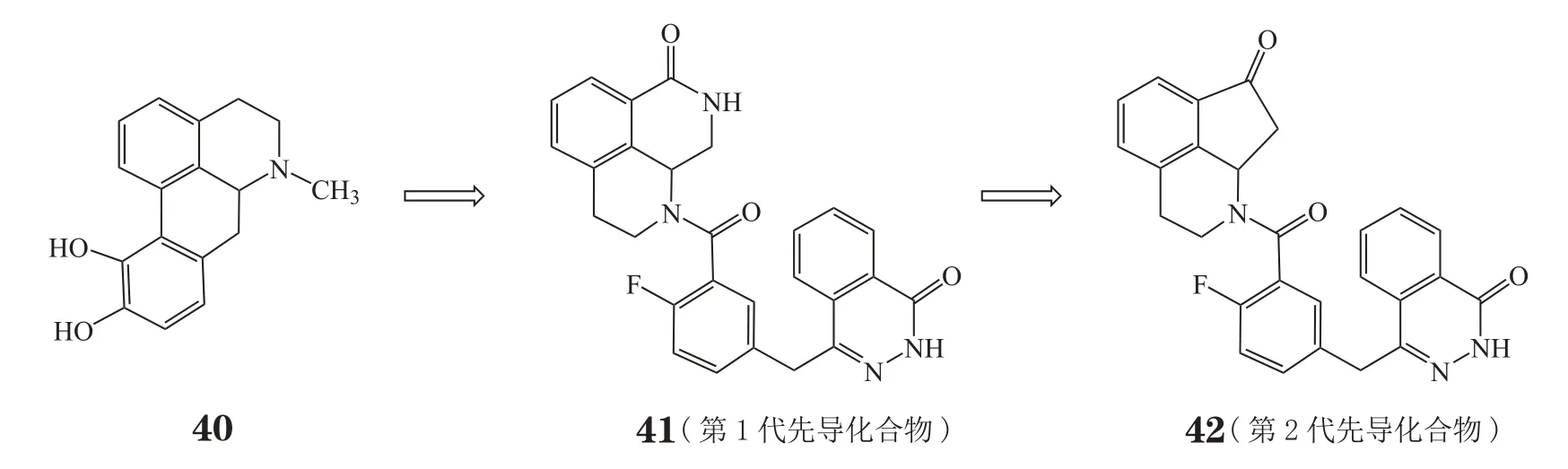
图16 以阿扑吗啡为起点的新型化合物优化过程Figure 16 Evolution of the novel PARP-1 inhibitor from apomorphine
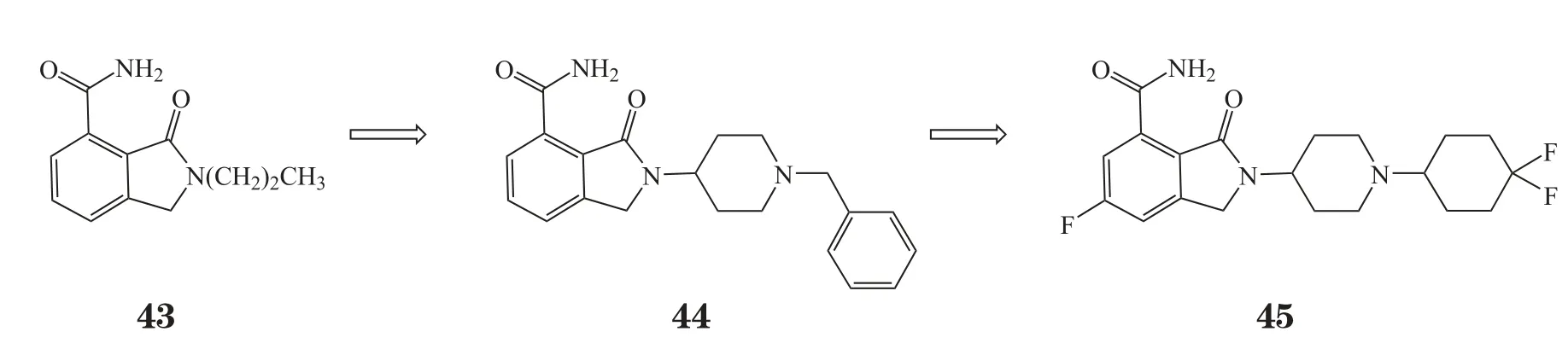
图17 NMS-P118的优化过程Figure 17 Evolution of NMS-P118
在临床前实验中,NMS-P118显示出很好的PARP-1选择性和较好的药动学性质,在小鼠和大鼠体内的口服生物利用度都很高,在BRCA1突变的MDA-MB-436细胞和BRCA2突变的Capan-1细胞中均显示出高单药活性和TMZ联用活性。作为第1个PARP-1特异性抑制剂,NMS-P118具有很强的临床开发潜力。
6 展望
随着分子医学的发展,肿瘤治疗已进入个体化治疗时代,PARP-1 抑制剂的显著抗肿瘤活性使其有着广阔的潜在应用研究空间。但同时大量实验证明,目前的大部分PARP-1抑制剂对PARP-1缺乏一定的特异性,且非所有BRCA突变的肿瘤细胞都对PARP-1抑制剂敏感[33],即易产生耐药性。耐药性产生机制可能有3种:1)BRCA功能的修复[34];2)Mdr1基因过度表达造成的P-糖蛋白对PARP抑制剂的消除(如奥拉帕尼即为P-糖蛋白的典型消除底物);3)HR功能的修复[18]。同时,PARP-1抑制剂对肿瘤具有选择性,对非BRCA突变肿瘤细胞的敏感缺乏特异性,导致目前进入临床的PARP-1抑制剂均未在试验中显示出可靠的化疗增敏效果,所有的Ⅲ期临床研究均以DNA修复缺陷的患者为对象。另外,由于其抑制了细胞中的DNA损伤修复,可能使正常细胞更易发生癌变,具有不可忽视的细胞毒性。就目前的临床候选药物来看,较晚进入临床的尼拉帕尼和BMN-673等具有较强的PARP-1捕获能力,与早期进入临床的化合物相比具有一定优势。因此,未来对PARP-1抑制剂的研究,应着力增强其特异性、改善细胞对其耐药性,重点挖掘其单药治疗DNA修复缺陷患者的潜力。同时,需要对PARP-1 抑制剂的作用特点和机制进行更深入的探索,对PARP-1参与DNA损伤修复的信号通路进行更全面的研究,以设计出更多新的PARP-1抑制剂,拓展其在肿瘤治疗中的应用。
[1]Qiu W, Lam R, Voytyuk O, et al. Insights into the binding of PARP inhibitors to the catalytic domain of human tankyrase-2[J]. Acta Crystallogr D Biol Crystallogr, 2014, 70(Pt 10): 2740-2753.
[2]Chen A. PARP inhibitors: its role in treatment of cancer[J]. Chin J Cancer, 2011, 30(7): 463-471.
[3]Langelier M F, Planck J L, Roy S, et al. Structural basis for DNA-dependent poly(ADP-ribosyl)ation by human PARP-1[J]. Science, 2012,336(6082): 728-732.
[4]Langelier M F, Servent K M, Rogers E E, et al. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation[J]. J Biol Chem, 2008, 283(7): 4105-4114.
[5]Lord C J, Ashworth A. The DNA damage response and cancer therapy[J]. Nature, 2012, 481(7381): 287-294.
[6]Rouleau M, Patel A, Hendzel M J, et al. PARP inhibition: PARP1 and beyond[J]. Nat Rev Cancer, 2010, 10(4): 293-301.
[7]Koh D W, Dawson T M, Dawson V L. Mediation of cell death by poly(ADP-ribose) polymerase-1[J]. Pharmacol Res, 2005, 52(1): 5-14.
[8]Curtin N J, Sharma R A. PARP Inhibitors for Cancer Therapy[M]. New York: Humana Press, 2015: 183-203.
[9]Wu Z, Wang C, Bai M, et al. An LRP16-containing preassembly complex contributes to NF-kappaB activation induced by DNA doublestrand breaks[J]. Nucleic Acids Res, 2015, 43(6): 3167-3179.
[10]Murai J, Huang S Y, Das B B, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors[J]. Cancer Res, 2012, 72(21): 5588-5599.
[11]Murai J, Huang S Y, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib[J]. Mol Cancer Ther, 2014, 13(2): 433-443.
[12]Bryant H E, Schultz N, Thomas H D, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase[J]. Nature, 2005, 434(7035): 913-917.
[13]Farmer H, McCabe N, Lord C J, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy[J]. Nature, 2005,434(7035): 917-921.
[14]Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside[J]. Annals of Oncology, 2011, 22(2): 268-279.
[15]Ferraris D V. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1)inhibitors. From concept to clinic[J]. J Med Chem, 2010, 53(12): 4561-4584.
[16]Menear K A, Adcock C, Boulter, R et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1[J]. J Med Chem, 2008, 51(20): 6581-6591.
[17]Menear K A, Adcock C, Boulter R, et al. Phthalazinones. Part 1: the design and synthesis of a novel series of potent inhibitors of poly(ADP-ribose) polymerase[J]. Bioorg Med Chem Lett, 2005, 15(9): 2235-2238.
[18]Jaspers J E, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors[J]. Cancer Discov, 2013, 3(1): 68-81.
[19]Canan Koch S S, Thoresen L H, Tikhe J G, et al. Novel tricyclic poly(ADP-ribose) polymerase-1 inhibitors with potent anticancer chemopotentiating activity: design, synthesis, and X-ray cocrystal structure[J]. J Med Chem, 2002, 45(23): 4961-4974.
[20]Wahlberg E, Karlberg T, Kouznetsova E, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors[J]. Nat Biotechnol, 2012, 30(3): 283-288.
[21]Chuang H C, Kapuriya N, Kulp S K, et al. Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triplenegative breast cancer cells[J]. Breast Cancer Res Treat, 2012, 134(2): 649-659.
[22]Sleijfer S, Bogaerts J, Siu L L. Designing transformative clinical trials in the cancer genome era[J]. J Clin Oncol, 2013, 31(15): 1834-1841.
[23]Liu J F, Konstantinopoulos P A, Matulonis U A. PARP inhibitors in ovarian cancer: current status and future promise[J]. Gynecol Oncol,2014, 133(2): 362-369.
[24]Penning T D, Zhu G D, Gandhi V B, et al. Discovery and SAR of 2-(1-propylpiperidin-4-yl)-1H-benzimidazole-4-carboxamide: A potent inhibitor of poly(ADP-ribose) polymerase (PARP) for the treatment of cancer[J]. Bioorg Med Chem, 2008, 16(14): 6965-6975.
[25]Penning T D, Zhu G D, Gandhi V B, et al. Discovery of the poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer[J]. J Med Chem, 2009, 52(2): 514-523.
[26]Jones P, Altamura S, Boueres J, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors[J]. J Med Chem, 2009, 52(22): 7170-7185.
[27]Sandhu S K, Schelman W R, Wilding G. The poly(ADP-ribose)polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial[J]. Lancet Oncol, 2013, 14(9): 882-892.
[28]Michie C O, Sandhu S K, Schelman W R, et al. Final results of the phase I trial of niraparib (MK4827), a poly(ADP)ribose polymerase(PARP) inhibitor incorporating proof of concept biomarker studies and expansion cohorts involving BRCA1/2 mutation carriers, sporadic ovarian, and castration resistant prostate cancer (CRPC)[J]. J Clin Oncol, 2013, 31: 2513.
[29]Shen Y, RehmanF L, FengY , et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency[J]. Clin Cancer Res, 2013, 19(18): 5003-5015.
[30]Zhang J, Lautar S, Huang S, et al. GPI 6150 prevents H2O2 cytotoxicity by inhibiting poly(ADP-ribose) polymerase[J]. Biochem Biophys Res Commun, 2000, 278(3): 590-598.
[31]Ye N, Chen C H, Chen T, et al. Design, synthesis and biological evaluation of a series of benzo[de][1,7]naphthyridin-7(8H)-ones bearing a functionalized longer chain appendage as novel PARP1 inhibitors[J]. J Med Chem, 2013, 56(7): 2885-2903.
[32]Papeo G, Posteri H, Borghi D, et al. Discovery of 2-[1-(4,4-difluorocyclohexyl)piperidin-4-yl]-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole-4-carboxamide (NMS-P118): a potent, orally available and highly selective PARP-1 inhibitor for cancer therapy[J]. J Med Chem, 2015,58(17): 6875-6898.
[33]Audeh M W, Carmichael J, Penson R T, et al. Oral poly(ADP-ribose)polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial[J]. Lancet, 2010, 376(9737): 245-251.
[34]Edwards S L, Brough R, Lord C J, et al. Resistance to therapy caused by intragenic deletion in BRCA2[J]. Nature, 2008, 451(7182): 1111-1115.
Progress in Research of Inhibitors Targeting PARP-1 for Cancer Therapy
XIE Yiyue, SHI Tianchen, WANG Luying, XU Xiaowei, LI Yanghui, LIN Kejiang
(School of Pharmacy, China Pharmaceutical University, Nanjing 210009,China)
Poly(ADP-ribose) polymerase-1(PARP-1) plays an important role in repairing DNA damage and represents a currently important target in cancer therapy. Based on the structure and mechanism of PARP-1, the major structure types of PARP-1 inhibitors and optimization strategies were reviewed, along with an outlook on their future applications and urgent problems to be solved.
PARP-1; inhibitor; cancer
R918
A
1001-5094(2015)10-0761-14
接受日期:2015-10-08
项目资助:国家自然科学基金(No.81573348)
*通讯作者:林克江,副教授;
研究方向:新药设计与研发;
Tel:025-83271455;E-mail:link@cpu.edu.cn
