E-cadherin在肿瘤治疗中的研究进展
2015-11-24常琳琳朱虹郑琳曹戟罗沛华何俏军
常琳琳,朱虹,郑琳,曹戟,罗沛华,何俏军
(浙江大学药学院 浙江省抗肿瘤药物临床前研究重点实验室,浙江 杭州 310058)
E-cadherin在肿瘤治疗中的研究进展
常琳琳,朱虹,郑琳,曹戟,罗沛华,何俏军*
(浙江大学药学院 浙江省抗肿瘤药物临床前研究重点实验室,浙江 杭州 310058)
E-cadherin 参与形成细胞间黏附性连接,是胚胎发育过程中的一个关键因子。越来越多的研究表明,E-cadherin在肿瘤的发生发展过程中也发挥了至关重要的作用。在生物体内,E-cadherin的表达和功能受到多个水平、多重因素的调控,而E-cadherin 又可以影响多条重要信号通路的活性,参与到多种生理病理过程中。E-cadherin下调造成细胞间黏附性连接减少、极性减弱,细胞由上皮样转变为间质样,这一变化是上皮间质转化(EMT)的重要标志之一。E-cadherin与多种肿瘤的发生有一定的相关性。同时E-cadherin下调所引起的EMT促进肿瘤细胞的迁移运动,肿瘤细胞侵袭力增强,促进转移的发生。近年来,大量研究关注到E-cadherin对肿瘤细胞的耐药及干细胞特性的获得都有影响。综述E-cadherin在肿瘤发生发展中的作用,探讨以E-cadherin为靶点的肿瘤治疗的现状及展望。
E-cadherin;上皮间质转化;肿瘤发生;转移;耐药;肿瘤干细胞
钙黏蛋白(cadherin)是一类细胞表面的跨膜糖蛋白,对细胞-细胞间的黏附功能有着重要影响。在钙黏蛋白超家族成员中,经典钙黏蛋白是最先被发现的,它是细胞间黏附性连接的重要结构组成。钙黏蛋白通过其细胞外结构域介导Ca2+依赖的、同源性细胞-细胞间黏附。钙黏蛋白将细胞外环境与细胞骨架相连接,并通过其保守的胞浆结构域与p120-、α-、β-连环蛋白(catenin)等连环蛋白的相互作用参与细胞信号的传导。Ⅰ类经典钙黏蛋白E-cadherin是上皮组织中构成细胞间黏附性连接的重要组成部分[1]。
人源性E-cadherin由CDH1基因编码,是目前研究最多的钙黏蛋白家族成员。它的胞外结构域包含5个钙黏蛋白重复(EC1-EC5)、1个跨膜结构域以及1个胞内结构域。E-cadherin的胞内结构域包含1个高磷酸化的区域,与β-catenin的结合密切相关,进而影响着E-cadherin的功能[2]。β-catenin也能够与α-catenin结合,α-catenin参与调控包含actin的细胞骨架形成。在上皮细胞中,包含E-cadherin的细胞间连接通常与包含actin的细胞骨架相毗邻。
E-cadherin参与调控上皮的形成、维持内稳态,其首要功能在于形成黏附性连接,这一作用对多种组织不同细胞中的细胞间隙及细胞相互作用的稳态都至关重要[3]。随着对E-cadherin关注的增加和研究的深入,E-cadherin在肿瘤的发生、发展及治疗中的作用和地位受到了越来越多的肯定。本文将在介绍E-cadherin的调控机制、相关信号通路的基础上,着重阐述E-cadherin与肿瘤发生、转移、耐药及肿瘤干细胞的关系,进一步展望其在肿瘤治疗过程的作用和前景。
1 E-cadherin的调控机制
E-cadherin在细胞内的表达和功能受到多种因素多水平的调控。包括转录调控、蛋白翻译后水平调控等。
1.1 转录因子调控
Rb、c-Myc、WT1等能够通过结合CDH1位于5端序列的结合位点,从而激活CDH1。另一个正性调控CDH1的调控因子是肝细胞核因子-3(hepatocyte nuclear factor-3,HNF3),它通过协同AML-1及p300激活E-cadherin的转录[4]。另一方面,CDH1受到E-pal盒(包含启动子的2个毗邻E盒)的失活的负性调控。Snail是首个被发现的通过结合E盒区作用的E-cadherin特异性转录抑制因子(Behrens等, Proc Natl Acad Sci USA,1991年)。Slug(也被称作SNAI2)、E12/E47、ZEB1及ZEB2等随后也陆续被证实是E-cadherin的负性调控因子[5]。这些转录抑制因子通过募集组蛋白去乙酰化酶(HDACs)及CtBP、mSinA等转录共抑制物来发挥作用[6]。
1.2 甲基化调控
CDH1启动子CpG岛的高甲基化是调控其基因表达的重要机制。Graff 研究团队于1995年在Cancer Res上首次报道,在正常上皮组织中CDH1启动子未甲基化,然而在多种肿瘤细胞中均发现了CDH1启动子的异常甲基化。K-Ras等基因能够通过影响E-cadherin启动子的甲基化水平,进而影响E-cadherin的表达[7]。
1.3 microRNA调控
MicroRNA也在CDH1基因表达过程中发挥了重要的调控作用。例如,miR-200 家族成员通过直接靶向E-cadherin的转录抑制因子ZEB1、ZEB2,调节E-cadherin的表达[8]。抑制内源性miR-200表达,其对ZEB1、ZEB2的抑制作用减弱,从而E-cadherin转录水平表达减少,最终引起细胞的间质样形态以及细胞迁移。除了miR-200家族,miR-9和miR-101也参与了E-cadherin的调控过程[9]。在乳腺癌细胞中,Myc诱导产生miR-9,后者直接靶向E-cadherin,随后激活β-catenin信号通路及VEGF表达,促进细胞运动、侵袭及血管生成。采用miR-9的抑制剂作用于细胞后,细胞中E-cadherin表达增加。Carvalho等[10]发现,在胃癌中miR-101缺失会引起E-cadherin的功能紊乱。
1.4 蛋白翻译后调控
除了以上调控方式之外,E-cadherin在细胞中的表达还受到蛋白翻译后修饰和调控。首先,细胞可通过胞吐运输方式,循环利用胞内体控制新合成的E-cadherin向细胞膜的传递。在此过程中,β-catenin与E-cadherin结合,两者以复合体的形式转运到细胞膜。
此外,细胞的内吞通路也调控E-cadherin的内吞,进而影响蛋白周转、循环、隔离及降解[9]。p120-catenin是最经典的E-cadherin内吞抑制剂,它可阻碍clathrin介导的E-cadherin内吞,并且能够稳定细胞膜表面的E-cadherin。蛋白激酶C等多种因素能够调节E-cadherin的内吞和循环,从而影响E-cadherin的功能。
磷酸化是E-cadherin的另一重要调控方式:E-cadherin/β-catenin复合体的酪氨酸磷酸化引起E-cadherin的内吞作用并且干扰连接复合体的稳定性。Src激活会增加E-cadherin的泛素化,随后E-cadherin经由溶酶体通路降解,而不是按照常规途径循环回细胞膜发挥其功能[11]。
另一个重要的E-cadherin 翻译后调控方式是其糖基化。E-cadherin 能够发生O-和N-糖基化,并且这2种修饰均能够影响E-cadherin 的功能[12]。新合成的E-cadherin发生细胞质O-糖基化(O-GlcNAc)会阻碍E-cadherin向细胞膜的转运,导致细胞间黏附减弱。而N-糖基化则是E-cadherin表达、折叠及运输所必需。人源E-cadherin的细胞外结构域在氨基酸基序上存在4个潜在的N-糖基化位点,N-乙酰氨基葡萄糖转移酶Ⅲ和Ⅴ(GnT-III和GnT-V)竞争性修饰E-cadherin N-聚糖,从而调节E-cadherin的膜表达。
2 E-cadherin相关信号通路
E-cadherin的表达、细胞定位及功能对细胞内多条信号通路都具有一定的影响,如:细胞支架组成、整合素、生长因子受体等相关信号通路。
E-cadherin对Wnt信号通路具有重要的调控作用,主要通过影响β-catenin的活性发挥作用。Wnt蛋白与Frizzled家族的细胞膜表面受体结合,从而介导β-catenin从细胞质转位至细胞核中,在胞核中激活T细胞因子(T-cell factor)/淋巴样增强因子(lymphoid enhancer factor)复合体,引起一系列靶基因的转录。其靶基因包括重要的癌基因c-Myc。Wnt通路在胚胎发育过程中具有重要作用,同时它还对细胞迁移、增殖、细胞间黏附等过程具有调控作用。而细胞质中的β-catenin水平受到细胞膜表面与E-cadherin结合形成黏附性连接的影响,干扰β-catenin与E-cadherin形成的黏附连接,能够影响细胞质中β-catenin的水平,进而影响Wnt通路。E-cadherin-β-catenin相关信号还可通过调节小GTP酶Rho家族成员的活性而影响细胞支架信号网络的调控。Rho小GTP酶超家族包括Rho、Rac和Cdc42等3个成员。其中,Rho有助于维持细胞微丝骨架的稳定或促进细胞骨架聚合,提高细胞收缩力,促进细胞迁移;Rac参与了细胞迁移过程中板状伪足的形成;Cdc42则诱导丝状伪足的形成,从而明确细胞运动的方向。E-cadherin外显子8读码框内缺失将导致Rac1激活减弱,并且抑制Rho的活性[13]。除了β-catenin之外,catenin家族的另一个重要分子p120也与E-cadherin关系密切。如前所述,p120与E-cadherin的胞内区段结合后,封闭其泛素化位点,从而阻断其内吞,维持细胞和细胞之间的紧密黏附。此外,通过与E-cadherin的结合,p120还可以调控Rho GTPase的活性,从而影响细胞骨架的结构及运动能力[14](见图1)。

图1 E-cadherin与β-catenin和p120的交互作用Figure 1 The interaction of E-cadherin with β-catenin and p120
另一方面,E-cadherins与受体酪氨酸激酶(RTKs)存在互相调控的关系,E-cadherin能抑制RTKs的激活,进而影响与细胞稳态密切相关RTKs介导的信号通路。特别是,有研究报道EGFR与E-cadherin存在双向的交互作用:E-cadherin参与的细胞黏附能够抑制EGF依赖的EGFR激活;然而在细胞间连接形成时,E-cadherin短暂性激活EGFR。E-cadherin干扰EGFR、c-Met及IGF-1R信号通路,主要是通过它们之间同源的受体,影响配体与受体之间的亲和力。E-cadherin与EGFR结合后阻断其下游信号通路,进而引起RhoA的激活,促进细胞运动(Takahashi等, Exp Cell Res, 1996年)。
研究表明E-cadherin的缺失与细胞的侵袭能力提高密切相关。与野生型细胞相比,E-cadherin突变的细胞内源性分泌MMP9增多。而MMP可裂解E-cadherin的胞外结构域,使E-cadherin失活。E-cadherin的胞外域水解片段会干扰E-cadherin/catenin复合体中E-cadherin的功能,促进细胞侵袭。此外,E-cadherin下调或功能缺失使得细胞对凋亡刺激更耐受。E-cadherin突变的细胞不仅具有更强的侵袭力,同时在Notch-1的介导作用下Bcl-2发生上调,细胞存活增加[15]。E-cadherin下调引起β-catenin在细胞核内累积,β-catenin/TCF反式激活,进而细胞PI3K/Akt信号通路活性上调,PI3K激活能够进一步稳定β-catenin通路,最终促进细胞生长。
3 E-cadherin与肿瘤的关系
随着针对E-cadherin的研究不断深入,E-cadherin与肿瘤的关系也受到了越来越多的关注。众多研究表明E-cadherin是一个抑癌因子,抑制E-cadherin的功能或表达将导致细胞形态向间质细胞样转化,促进细胞的迁移、侵袭以及转移。E-cadherin的下调造成细胞基底膜的越界和蔓延,促进某些上皮衍生的肿瘤的早期侵袭性行为。反之,在缺失E-cadherin的细胞株中重新转入E-cadherin能够逆转细胞的低分化肿瘤表型(例如:成纤维细胞样、高侵袭力、细胞间黏附小等),使得细胞恢复到低侵袭力上皮样表型、细胞间黏附紧密的高分化状态[16]。E-cadherin的减少能够激活多条致瘤的信号通路,包括MAPK、PI3K、NF-κB通路等[17]。上述信号通路都与细胞的多个生理过程密切相关,E-cadherin的表达或功能还能影响肿瘤细胞对抗肿瘤治疗的敏感性。此外,近年来越来越多的研究关注到E-cadherin与肿瘤细胞的干细胞特征也有一定的关联。
3.1 E-cadherin、上皮间质转化与肿瘤转移
在晚期肿瘤中,E-cadherin表达或其细胞膜定位往往发生缺失,并且这一变化与肿瘤转移和复发的高发生率密切相关。在乳腺癌、胃癌等人肿瘤模型中,E-cadherin编码基因CDH1的缺失或异常表达会导致肿瘤细胞侵袭周围的组织或器官。这主要是通过上皮间质转化(epithelial-to-mesenchymal transition,EMT)实现的。极化的上皮细胞经历多种生化变化后转化为间质细胞的表型,细胞间黏附减少,上皮细胞蛋白也随之减少,其中最主要的就是E-cadherin[18]。上皮来源的肿瘤细胞中,E-cadherin参与的细胞黏附性连接相对紧密,同时细胞与细胞外基质的连接也比较牢固,这些特征不利于肿瘤细胞发生转移。而肿瘤细胞经历EMT后,随着各种黏附性连接的减弱,细胞被赋予更强的迁移侵袭能力,能够从原发灶迁移进入循环系统,进而在新的组织器官形成转移灶。
肿瘤转移是临床上肿瘤病人最主要的致死原因。临床肿瘤转移发生率高,并且对肿瘤患者预后造成严重影响,因此亟需全新的治疗策略来预防高转移风险病人的肿瘤从某一局限器官扩散或者消除晚期患者已经存在的转移细胞。Hedgehog通路抑制剂cyclopamine(1)作用于胰腺癌细胞后,抑制Snail表达,上调E-cadherin水平,尤其针对一些已具有EMT初始特征的细胞,能够显著地抑制肿瘤转移[19]。
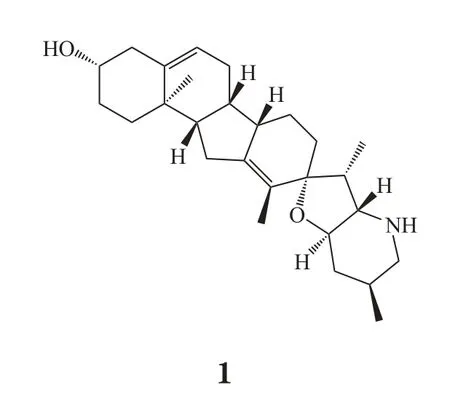
目前在肿瘤转移的预防/治疗手段中,直接靶向E-cadherin或EMT过程中一些关键结构蛋白及转录因子的治疗方式相对匮乏,主要是通过抑制EMT的诱导因素。细胞外诱导EMT发生的刺激因素的拮抗剂或者抑制EMT相关信号传导通路中的关键节点被视为抑制EMT发生的有效策略之一。肿瘤微环境中多种信号刺激均能够诱导EMT的发生,包括氧含量降低、细胞因子及多种生长因子等。针对上述特点,靶向这些细胞外信号在细胞表面的特异性受体可能能够有效地抑制肿瘤细胞发生EMT。但由于肿瘤微环境中引起EMT的信号复杂多样,如果单一地抑制某一受体活性,而其他EMT诱导因子不受影响,在治疗过程中很容易出现耐药,因此该策略具有明显的局限性,也限制了其临床应用。针对上述策略的局限性,靶向引起EMT的共同的关键信号转导通路可能能够克服抑制单一受体带来的问题[20]。例如,转录因子STAT3在多种人类肿瘤的EMT发生过程中都扮演了重要的角色[21]。STAT3抑制剂可能有望成为抑制EMT发生的有效策略。此外,靶向钙离子信号通路等参与EMT调控的上游信号通路也是可选的策略。无论是表皮生长因子(EGF)还是低氧诱导的EMT,都需要细胞内钙离子信号通路的参与,而TRPM7通道就可能成为钙离子依赖的STAT3激活引起的EMT的治疗靶点之一[22]。不过,靶向EMT诱导因子仅适合作为预防转移或治疗早期肿瘤转移的策略,因此更适合作为辅助治疗。因为一旦转移已经发生,肿瘤细胞已经经历了EMT并处于间质状态,这种情况下如果仅仅抑制EMT的诱导因子,患者很难从中获益。EMT是细胞获得间质细胞的特征的过程,在该过程中,间质标志物如vimentin、N-cadherin 及fibronectin等发生显著上调。这些间质标记物与细胞侵袭密切相关,并且通常表达于循环系统中的肿瘤细胞及转移灶[23]。因此,靶向间质性蛋白的治疗方案可能能够消除或改善肿瘤患者已经发生的转移细胞。这个策略在靶向侵袭性、间质样肿瘤细胞的同时,还可能对间质来源的肿瘤(肉瘤等)具有一定的疗效。
3.2 E-cadherin与肿瘤耐药
E-cadherin在影响肿瘤的转移、侵袭、浸润的同时,其下调也与肿瘤细胞对化疗、放疗等肿瘤治疗的耐药有着密不可分的关系。众多的研究表明,肿瘤细胞发生EMT后,不仅具有更强的侵袭力,同时还更容易对抗肿瘤治疗产生耐药。TGF-β及EGF在诱导EMT发生的同时,还能引起非小细胞肺癌细胞对顺铂、紫杉醇的耐药[24]。在乳腺癌细胞中也观察到5-FU对肿瘤细胞杀伤力随着一些间质标记物的增加而减弱[25]。除了传统化疗药物,分子靶向药物的抗肿瘤活性也与细胞的EMT具有密切的关联。高表达间质标记物vimentin、fibronectin的肺癌细胞,在体内外均表现出对EGFR抑制剂(如厄洛替尼、吉非替尼、西妥昔单抗)的敏感性较低[26]。
然而,E-cadherin引起抗肿瘤药物耐药的机制尚不明确,研究发现肿瘤细胞敲除E-cadherin后,细胞的增殖能力增强,这可能在一定程度上能解释E-cadherin下调对耐药的影响。此外,E-cadherin的下调可以间接激活PI3K通路以及NF-κB通路,而这2条通路与肿瘤的发生、发展以及耐药密切相关,这可能是E-cadherin引起耐药的一部分原因。更值得注意的是,E-cadherin的下调会造成肿瘤细胞表现出很多肿瘤干细胞样特性,而肿瘤干细胞往往更容易产生耐药性。
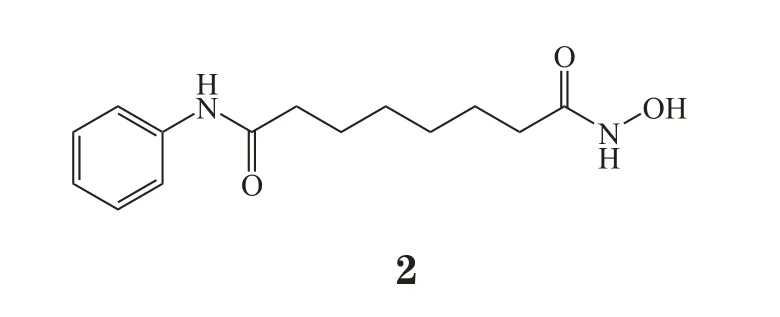
不过,E-cadherin下调与肿瘤耐药的相关性在带来困扰的同时,也给肿瘤治疗提供了全新的思路。一方面,提示在肿瘤治疗过程中根据E-cadherin表达水平选择合适的方案。针对E-cadherin缺失/低表达的患者,E-cadherin应该作为选择治疗方案时的考量标准之一。例如,在临床研究中发现,E-cadherin高表达的非小细胞肺癌患者采用厄洛替尼治疗后,疗效普遍较好;反之,E-cadherin阴性患者在接受厄洛替尼治疗后,病情依然进一步恶化。另一方面,基于E-cadherin调控的联合用药也受到了极大的关注。在乳腺癌细胞MCF-7中诱导EMT发生,细胞产生对阿霉素的耐药,敲除Twist和ZEB1能够逆转EMT并克服耐药。值得关注的是,HDAC抑制剂能够促进E-cadherin的转录,从而增加细胞内E-cadherin的表达,采用HDAC抑制剂与其他抗肿瘤药物联合用药,对于克服E-cadherin引起的耐药有较高的可行性。HDAC抑制剂SAHA(2)与吉非替尼合用,在头颈部鳞状细胞癌中表现出了很好的协同作用[27]。此外,天然来源的抗肿瘤化合物雷公藤红素的抗肿瘤活性也与E-cadherin表达相关,而SAHA与雷公藤红素在多种实体瘤细胞中实现了协同抗肿瘤作用,并且在裸小鼠荷瘤模型中也得到了验证[28]。
3.3 E-cadherin与肿瘤干细胞
肿瘤干细胞(cancer stem cell,CSC)是指存在于肿瘤细胞中的一小部分具有干细胞特性的细胞群。目前研究者普遍认为CSC是肿瘤转移、耐药、预后不良及复发的主要原因。从正常乳腺组织或者乳腺癌组织中分离得到的干细胞,表现出E-cadherin下调等一系列典型的EMT样指征变化。在乳腺癌肿瘤干细胞中,E-cadherin显著下调,E-cadherin 的这种变化促进肿瘤生成以及参与低氧耐药的产生。在肺癌病例中,E-cadherin下调引起的EGFR抑制剂耐药现象也与肿瘤干细胞样特性相关[29]。
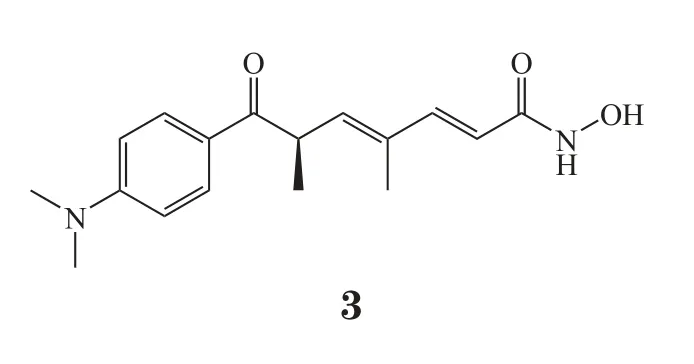
越来越多的研究都显示E-cadherin的下调及EMT的发生与CSC存在密切的相关性。考虑到CSC在肿瘤发生发展中的作用和地位,以及CSC与普通肿瘤细胞的差异,肿瘤治疗中针对CSC的策略显得尤为重要。首先受到关注的就是靶向CSC特有的细胞表面标记物,如CD133、CD44等,主要是采用抗体来发挥作用[30]。另外,有研究者对候选化合物进行筛选,挑选出具有CSC杀伤作用的抗肿瘤化合物。其中,抑制HDAC被认为是诱导间质样肿瘤细胞及CSC分化的有效策略,能够诱导凋亡或增敏其他抗肿瘤药物。目前,trichostatin A(3)及SAHA等HDAC抑制剂单用或与其他药物联合用药等多个方案均处于临床研究阶段[28-32]。此外,糖尿病治疗药物AMPK激动剂二甲双胍与其他化疗药物合用,也表现出杀伤CSC的抗肿瘤活性[33-34]。由于EMT与CSC的相关性,上文中讨论到的抑制EMT的策略同时靶向CSC的可能性很大,比如干扰ZEB1的表达、抑制TGF-β等。PI3K/PTEN/mTOR信号通路在肿瘤耐药及CSC中均发挥了突出的作用[35]。研究表明,PI3K活性与CSC的扩增和维持直接相关。抑制PI3K/Akt/ mTOR通路活性能够阻断CSC的自我更新,并且克服CSC引起的肿瘤耐药[36]。
4 总结与展望
自从E-cadherin 被发现以来,随着科学家对其关注的增加以及研究的深入,E-cadherin的生物学功能和病理学意义不断被拓展,E-cadherin参与调控的生命过程错综复杂。E-cadherin在发育过程中的作用和地位被进一步巩固,同时它在肿瘤的发生发展过程中所扮演的角色也越来越突出。因此,基于E-cadherin调控的肿瘤治疗手段具有十分重要的理论和实践意义。编码E-cadherin的基因CDH1突变或缺失会引起多种肿瘤的发生率显著提高,检测CDH1有利于预警某些肿瘤的发生可能。
E-cadherin 缺失或下调引起肿瘤细胞发生EMT,使得肿瘤细胞的迁移侵袭能力增强,促进肿瘤转移的发生。肿瘤转移是临床上肿瘤患者最主要的致死原因,针对E-cadherin及EMT的肿瘤治疗手段能够有效地预防和改善肿瘤转移,提高肿瘤患者生存率。此外,E-cadherin的表达与肿瘤细胞的耐药产生密切相关,E-cadherin表达低的肿瘤细胞往往对某些药物或(和)放疗等治疗手段耐药。根据E-cadherin的表达水平合理选择肿瘤治疗方案,或者采用联合用药方式增加E-cadherin表达,均可能提高肿瘤细胞对治疗的响应程度,增加治疗的有效率。更重要的是,多项研究证明E-cadherin影响了肿瘤细胞的干细胞特性,而肿瘤干细胞是肿瘤转移、耐药、复发等多种恶性表征的元凶。
综上所述,基于E-cadherin调控的治疗手段,在肿瘤治疗中能从多个层面发挥作用,提高有效率、扼制转移、防止复发、改善预后等。不过,研究者依然面对不少困扰,E-cadherin在肿瘤中作用尚存在一些争议,同时调控E-cadherin相关信号通路的治疗手段的研发刚刚起步,普遍处于临床前研究阶段。尽管如此,可以预见的是,对E-cadherin在肿瘤发生发展中的作用进行深入全面的剖析,对于肿瘤治疗具有推动作用;而以E-cadherin为中心的诊断和治疗策略将具有良好的开发意义和应用前景。
[1]Pieters T, van Roy F. Role of cell-cell adhesion complexes in embryonic stem cell biology[J]. J Cell Sci, 2014 ,127: 2603-2613.
[2]van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer[J].Nat Rev Cancer, 2014 ,14(2):121-134.
[3]Bhatt T, Rizvi A, Batta S P, et al. Signaling and mechanical roles of E-cadherin[J].Cell Commun Adhes, 2013, 20(6):189-199.
[4]Liu Y N, Lee W W, Wang C Y, et al. Regulatory mechanisms controlling human E-cadherin gene expression[J]. Oncogene, 2005, 24(56): 8277-8290.
[5]Dong C, Wu Y, Yao J, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer [J].J Clin Invest, 2012, 122(4):1469-1486.
[6]Wong T S, Gao W, Chan J Y. Transcription regulation of E-cadherin by zinc finger E-box binding homeobox proteins in solid tumors [J].Biomed Res Int, 2014, 2014:921564.
[7]Abudukadeer A, Bakry R, Goebel G, et al. Clinical relevance of CDH1 and CDH13 DNA-methylation in serum of cervical cancer patients [J]. Int J Mol Sci, 2012, 13(7):8353-8363.
[8]Park S M, Gaur A B, Lengyel E, et al. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2[J]. Genes Dev, 2008, 22(7): 894-907.
[9]de Beco S, Amblard F, Coscoy S. New insights into the regulation of E-cadherin distribution by endocytosis[J]. Int Rev Cell Mol Biol, 2012, 295:63-108.
[10]Carvalho J, van Grieken N C, Pereira P M, et al. Lack of microRNA-101 causes E-cadherin functional deregulation through EZH2 up-regulation in intestinal gastric cancer[J]. J Pathol, 2012, 228(1): 31-44.
[11]Palacios F, Tushir J S, Fujita Y, et al. Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions[J]. Mol Cell Biol, 2005, 25(1): 389-402.
[12]Pinho S S, Seruca R, Gartner F, et al. Modulation of E-cadherin function and dysfunction by N-glycosylation[J]. Cell Mol Life Sci, 2011, 68(6): 1011-1020.
[13]Canel M, Serrels A, Frame M C, et al. E-cadherin-integrin crosstalk in cancer invasion and metastasis[J]. J Cell Sci, 2013, 126(Pt 2):393-401.[14]Leckband D E, de Rooij J. Cadherin adhesion and mechanotransduction[J]. Annu Rev Cell Dev Biol, 2014, 30:291-315.
[15]Ferreira A C, Suriano G, Mendes N,et al. E-cadherin impairment increases cell survival through Notch-dependent upregulation of Bcl-2[J]. Hum Mol Genet, 2012, 21(2): 334-343.
[16]Canel M, Serrels A, Frame M C, et al. E-cadherin-integrin crosstalk in cancer invasion and metastasis[J].J Cell Sci, 2013,126(Pt 2):393-401.
[17]Brouxhon S M, Kyrkanides S, Teng X, et al. Soluble E-cadherin: a critical oncogene modulating receptor tyrosine kinases, MAPK and PI3K/Akt/mTOR signaling[J]. Oncogene, 2014, 33(2): 225-235.
[18]Kalluri R, Weinberg R A. The basics of epithelial-mesenchymal transition[J]. J Clin Invest, 2009, 119(6): 1420-1428.
[19]Feldmann G, Fendrich V, McGovern K, et al. An orally bioavailable smallmolecule inhibitor of Hedgehog signaling inhibits tumor initiation andmetastasis in pancreatic cancer[J]. Mol Cancer Ther, 2008, 7(9): 2725-2735.
[20]Yu M, Bardia A, Wittner B S, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition[J]. Science, 2013, 339(6119): 580-584.
[21]Balanis N, Wendt M K, Schiemann B J, et al. Epithelial to mesenchymal transition promotes breast cancer progression via a fibronectin-dependent STAT3 signaling pathway[J]. J Biol Chem, 2013, 288(25): 17954-17967.
[22]Davis F M, Azimi I, Faville R A, et al. Induction of epithelialmesenchymal transition (EMT) in breast cancer cells is calcium signal dependent[J]. Oncogene, 2014, 33(18): 2307-2316.
[23]Bednarz-Knoll N, Alix-Panabieres C, Pantel K. Plasticity of disseminating cancer cells in patients with epithelial malignancies[J]. Cancer Metastasis Rev, 2012, 31(3/4): 673-687.
[24]Shintani Y, Okimura A, Sato K, et al. Epithelial to mesenchymal transition is a determinant of sensitivity to chemoradiotherapy in nonsmall cell lung cancer[J]. Ann Thorac Surg, 2011, 92(5): 1794-1804.
[25]Zhang W, Feng M, Zheng G,et al. Chemoresistance to 5-fluorouracil induces epithelial-mesenchymal transition viaup-regulation of Snail in MCF7 human breast cancer cells[J]. Biochem Biophys Res Commun,2012, 417(2): 679-685.
[26]Fuchs B C, Fujii T, Dorfman J D, et al. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells[J]. Cancer Res, 2008, 68(7): 2391-2399.
[27]Bruzzese F, Leone A, Rocco M, et al. HDAC inhibitor vorinostat enhances the antitumor effect of gefitinib in squamous cell carcinoma of head and neck by modulating ErbB receptor expression and reverting EMT[J]. J Cell Physiol, 2011, 226(9): 2378-2390.
[28]Zheng L, Fu Y, Zhuang L,et al. Simultaneous NF-kappaB inhibition and E-cadherin upregulation mediate mutually synergistic anticancer activity of celastrol and SAHA in vitro and in vivo[J]. Int J Cancer, 2014, 135(7): 1721-1732.
[29]Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells[J]. Cancer Res, 2013, 73(10): 3051-3061.
[30]Deonarain M P, Kousparou C A, Epenetos A A. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy?[J]. MAbs, 2009,1(1): 12-25.
[31]Walter R B, Medeiros B C, Gardner K M, et al. Gemtuzumab ozogamicin in combination with vorinostat and azacitidine in older patients with relapsed or refractory acute myeloid leukemia: a phase I/II study[J]. Haematologica, 2014, 99(1): 54-59.
[32]Holkova B, Supko J G, Ames M M, et al. A phase I trial of vorinostat and alvocidib in patients with relapsed, refractory, or poor prognosis acute leukemia, or refractory anemia with excess blasts-2[J]. Clin Cancer Res, 2013, 19(7): 1873-1883.
[33]Janzer A, German N J, Gonzalez-Herrera K N, et al. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells[J]. Proc Natl Acad Sci USA, 2014, 111(29): 10574-10579.
[34]Reddi A, Powers M A, Dellavalle R P. Therapeutic potential of the anti-diabetic agent metformin in targeting the skin cancer stem cell diaspora[J]. Exp Dermatol, 2014, 23(5): 345-346.
[35]Chang L, Graham P H, Hao J, et al. Acquisition of epithelialmesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance[J]. Cell Death Dis, 2013, 4: e875.
[36]Airiau K, Mahon F X, Josselin M, et al. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor[J]. Cell Death Dis, 2013, 4: e827.

[专家介绍] 何俏军 :博士,2005年毕业于浙江大学,美国南加州大学博士后。目前为浙江大学药学院教授、博导,教育部新世纪优秀人才,任浙江大学药理毒理研究所所长和浙江大学药物安全评价研究中心主任。研究领域主要包括:关键蛋白的翻译后调控模式在肿瘤分化及药物毒性过程中发挥的作用及相关信号转导通路,并基于此发现调控分化和防治毒性的新靶点和新型小分子药物。主持国家自然科学基金面上项目和重大研究计划项目、国家重大新药创制专项等国家级科研项目7项,在FASEB J、Cancer Research、Molecular Cancer Therapeutics等国际知名刊物发表SCI论文120余篇,获授权发明专利19项。获教育部科技进步二等奖、浙江省科技进步二等奖、中国专利优胜奖等奖项。任CFDA药品评审中心新药评审专家和中国毒理学会理事等职。
Research Progress of E-cadherin in Cancer Therapy
CHANG Linlin, ZHU Hong, ZHENG Lin, CAO Ji, LUO Peihua, HE Qiaojun
(Zhejiang Province Key Laboratory of Anti-Cancer Drug Research, School of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China)
E-cadherin; epithelial-mesenchymal transition; carcinogenesis; metastasis; drug resistance; cancer stem cell
R966;R962
A
1001-5094(2015)10-0754-07[Abstract] E-cadherin, an intercellular adhesion molecule, plays critical roles during embryonic development. Mounting evidences have revealed that E-cadherin is also involved extensively in the development and progression of cancer. E-cadherin expression and function are tightly controlled by multiple factors at multiple levels. Furthermore, E-cadherin is capable of regulating multiple important signaling pathways and involved in a variety of physiological and pathological processes. The downregulation of E-cadherin results in less intercellular adhesion and reduced cell polarity. Consequently, epithelial cells become mesenchymal stem cells, which is one of the hallmarks of epithelial-mesenchymal transition (EMT). Thus it has been proposed the correlation between the expression of E-cadherin and the malignance of tumors. E-cadherin downregulation-induced EMT facilitates the migration, invasion and metastasis of tumor cells. In addition, recent evidences have suggested that E-cadherin reduction also leads to drug resistance and increased stem cell properties of tumor cells. Based on these studies, the current review summarizes the roles of E-cadherin in carcinogenesis and cancer progression, and explores the current situation and future prospects of cancer therapy targeting E-cadherin.
接受日期:2015-10-08
项目资助:浙江省卫生厅高层次创新人才基金
*通讯作者:何俏军,教授,博士生导师;
研究方向:肿瘤药理学,药物毒理学;
Tel:0571-88208400; E-mail:qiaojunhe@zju.edu.cn
