CDK抑制剂在抗肿瘤领域的研发进展
2015-11-24谢韶丁健陈奕
谢韶,丁健,陈奕
(中国科学院上海药物研究所新药研究国家重点实验室肿瘤药理组,上海 201203)
CDK抑制剂在抗肿瘤领域的研发进展
谢韶,丁健,陈奕*
(中国科学院上海药物研究所新药研究国家重点实验室肿瘤药理组,上海 201203)
细胞周期蛋白依赖性激酶(cyclin dependent kinase,CDK)为细胞周期调节的关键激酶,参与细胞增殖、转录、存活等生理过程。CDK在多种肿瘤中异常活化,是抗肿瘤药物研发的重要靶点之一。目前已有1个CDK抑制剂(palbociclib, CDK4/CDK6抑制剂)被美国食品药品监督管理局批准于2015年上市,数十个CDK抑制剂处于针对实体瘤和血液系统肿瘤的临床或临床前研究阶段。综述目前抗肿瘤领域CDK抑制剂的研发现状、遇到的问题和可能的解决方案,并讨论其临床应用的可能。
CDK;抑制剂;ATP竞争;非ATP竞争;联合用药
过度活化、持续的细胞增殖是肿瘤的一个基本特征,因此诱导细胞周期阻滞可有效抑制肿瘤的生长。细胞周期蛋白依赖性激酶(cyclin dependent kinase,CDK)属于丝/苏氨酸蛋白激酶家族,是参与细胞周期调节的关键激酶。目前已报道有20个不同的CDK,它们都含有一段PSTAIRE的同源序列,并通过该序列与相应的调节亚基——细胞周期蛋白(cyclin)结合形成有活性的异源二聚体,参与转录、代谢、神经分化和发育等生理过程[1-4]。
1 CDK在肿瘤细胞中的作用
根据CDK功能的不同,可以将其主要分为两大类。一类CDK参与细胞周期调控,主要包括CDK1、CDK2、CDK4、CDK6等(见图1)。无外界信号刺激下,视网膜母细胞瘤蛋白(retinoblastoma protein, RB)与转录因子E2F结合并抑制其活性,抑制增殖,使细胞处于G0期。在外界有丝分裂信号刺激下,细胞进入G1期,cyclin D合成增加,与CDK4/6结合,使RB磷酸化,部分解除其对E2F的抑制,使周期进行所需蛋白cyclin E等表达增加。G1后期,cyclin E与CDK2结合,进一步磷酸化RB,E2F的活性被完全释放,细胞通过G1/S限制点,进入S期。此时,cyclin A取代cyclin E与CDK2结合,参与DNA复制。S后期,cyclin A与CDK1形成复合物,细胞进入G2期。此时,cyclin B与CDK1结合,驱动有丝分裂进行,促进细胞通过M期,最终完成细胞的有丝分裂过程。
另一大类CDK参与转录调节,主要包括CDK7、CDK8、CDK9、CDK10、CDK11等(见图1)。CDK7/ cyclin H、CDK8/cyclin C、CDK9/cyclin T均通过调节RNA聚合酶Ⅱ的磷酸化来调节转录。其中CDK7/cyclin H还组成CDK活化激酶(CDK activating kinase,CAK),调节CDK的活性。CDK9/cyclin T参与HIV的复制[5]。CDK3/cyclin C可调节RB磷酸化,参与G0/G1过渡,也能磷酸化活化复制因子1(activatingtranscription factor 1,ATF1),提高其转活化水平。CDK10/cyclin M通过磷酸化转录因子Ets来调节转录[6]。而CDK11/cyclin L在RNA剪接中发挥作用(Hu等,J Biol Chem,2003年)。
此外,CDK5则与传统意义上的CDK不同,其调节亚基不是cyclin,而是p35和p39,最初认为其只在神经系统中发挥作用,近期研究显示,CDK5也参与肿瘤发生发展相关的增殖、存活及上皮间充质转换等多个过程[7]。
在哺乳动物细胞中,CDK单体没有活性,只有与相应的调节亚基cyclin结合后才能变成有活性的稳定构象。细胞内CDK表达一般恒定,但cyclin却在细胞周期的不同阶段周期性地表达和降解,cyclin的这种表达模式决定了有序地活化不同CDK。Cyclin调节CDK的亚细胞定位,并通过特异的锚定位点来识别特异性的底物,发挥磷酸化功能[4]。此外,CDK/cyclin的活化也受到内源性抑制子抑制、活化及抑制磷酸化的调节。CDK内源抑制子(CDK inhibitor,CDKI)包括INK4(p15INKb、p16INKa、p18INKc、p19INKd)和CIP/KIP(p21cip1/waf1、p27Kip1、p57Kip2)两大家族,它们通过结合到CDK或CDK/cyclin复合物上,限制其构象改变,抑制酶活。CDK核苷酸结合口袋处的残基(如CDK1的T14、CDK2的T15)可被Wee1和Myt1磷酸化,阻止ATP结合,而这些抑制性磷酸化可被CDC25磷酸酶去磷酸化。CAK则使得CDK的T-loop(如CDK1的T161、CDK2的T160)发生活化磷酸化。
肿瘤通常被认为是由一群增殖异常活跃的细胞所构成。在肿瘤细胞中,cyclin过表达或过度活化、CDKI活性被抑制、上游分裂信号持续激活等都会引起CDK的活性改变。CDK活性失调会直接或间接引起细胞增殖失控、基因组不稳定(DNA突变增加,染色体缺失等)和染色体不稳定(染色体数目变化)等,参与肿瘤的发生发展[8]。由于CDK活性为细胞分裂所必需,而在肿瘤细胞中又常有CDK活性增强,因此长期以来,CDK一直被认为是抗肿瘤及其他增殖失调疾病药物研发的较好靶点。目前已有1个CDK抑制剂在临床用于抗肿瘤治疗,另有数十个CDK抑制剂正进行临床或临床前研究[3]。这些CDK抑制剂按作用机制不同,大致可分为ATP竞争性和非竞争性抑制剂[9]。
2 ATP竞争性CDK抑制剂
ATP竞争性CDK抑制剂是CDK抑制剂研发中进展顺利的一类,多为平面杂芳烃结构,由杂环家族组成或衍生而来,也有的来源于天然产物[2]。截至目前,已有一个该类抑制剂palbociclib被美国FDA批准进入临床使用,有近20个抑制剂处于临床研究阶段[10](见表1)。其中,flavopiridol(1)、roscovitine(2)等属于第1代进入临床的CDK抑制剂。这些抑制剂均模仿ATP结构以结合到CDK蛋白的ATP结合口袋而发挥抑制作用。由于不同CDK之间ATP口袋的氨基酸序列高度保守,尽管有些抑制剂有或高或低的CDK选择性,但大都缺乏CDK家族的选择性,因而在临床使用中出现较大的毒副作用[2]。因此,CDK抑制剂研发随后倾向于提高CDK的选择性和抗肿瘤能力。第2代CDK抑制剂的抗肿瘤活性和CDK相对选择性均更好,其中dinaciclib(7)和palbociclib(12)是目前研究最多的第2代CDK抑制剂。
2.1 CDK非选择性抑制剂
2.1.1 Flavopiridol Flavopiridol(alvocidib,1)为flavonoid半合成衍生物,由法国赛诺菲-安万特(Sanofi-Aventis)公司和美国国家癌症研究所(National Cancer Institute,NCI)联合开发,为首个进入临床实验的CDK抑制剂。Flavopiridol能显著抑制参与周期调节的CDK1、CDK2、CDK4、CDK6和转录调节的CDK7、CDK9的活性,IC50分别为30、170、100、60、300和10 nmol·L-1[2]。虽然其体外活性很好,但体内效果并不显著,在针对实体瘤的Ⅱ期临床研究中效果甚微(Schwartz等,J clin oncol,2001年)。但多个临床研究结果显示,它对造血系统恶性疾病如CLL、MCL效果较好[11]。在Ⅰ、Ⅱ期临床研究中显示出对CLL的显著疗效。在Ⅰ期临床研究中,有40%的病人对flavopiridol的治疗有反应[12];后续Ⅱ期研究中,对病情严重且前期使用其他治疗方案失败的CLL患者其有效率仍大于50%[13]。然而,其毒副作用强,需缓慢静脉滴注给药,限制了其临床应用[14]。Flavopiridol与其他多种抗肿瘤药物如多西他赛(docetaxel)、伊立替康(irinotecan)或顺铂(cisplatin)等均具有协同效果,这些联合用药能有效降低flavopiridol的使用剂量,从而减轻其毒副作用[15]。
2.1.2 R-Roscovitine R-Roscovitine(selicilib、CYC-202, 2)为嘌呤类CDK抑制剂,可口服,2001年由美国Cyclacel Pharmaceuticals推入临床研究,为第2个进入临床实验的CDK抑制剂。R-Roscovitine可抑制CDK1、CDK2、CDK5、CDK7、CDK9的活性,IC50分别为330、220、270、800和230 nmol·L-1[2,16]。其可显著抑制转录,下调抗凋亡蛋白Mcl-1的表达,从而诱导人类白血病和多发性骨髓瘤细胞发生凋亡[17]。在与吉西他滨(gemcitabine)和顺铂联用、或与多西他赛(docetaxel)联用治疗NSCLC的Ⅰ期临床研究中,口服R-roscovitine显示出较好的生物利用度[10]。R-Roscovitine单药治疗鼻咽癌的Ⅱ期临床研究中,有病人显示出显著的肿瘤消退[10,18]。但该化合物的临床研究进展似乎并不顺利,目前关于 R-roscovitine仅有一项针对库欣病的Ⅱ期临床研究。
2.1.3 Dinaciclib Dinaciclib(SCH-727965,MK-7965,7)由德国默克(Merck)公司开发,是活性非常强的CDK抑制剂,其对CDK1、CDK2、CDK5、CDK9均有抑制活性,IC50分别为3、1、1和4 nmol·L-1,而对CDK4、CDK6、CDK7的抑制活性较弱[19]。虽然该化合物对CDK的选择性较差,但它对CDK以外的其他激酶几乎没有抑制活性,与flavopiridol相比,靶点选择性相对有所改善[19]。最初dinaciclib在Ⅰ期临床研究中有较好的表现,能使多种恶性肿瘤病情无进展,且毒性可耐受,但进一步在实体瘤中的随机实验结果却令人失望[20]。然而,针对复发MM的Ⅰ、Ⅱ期临床研究初步结果显示,dinaciclib单药即有一定疗效,27人中有2人有部分反应[21]。与flavopiridol一样,dinaciclib治疗CLL效果较好。2012年,dinaciclib进入针对CLL治疗的Ⅲ期临床研究,该研究比较了dinaciclib与CD20单抗奥法木单抗(ofatumumab)在氟达拉滨(fludarabine)或化学免疫治疗耐受CLL病人的疗效[22],2014年底已完成,但目前还未有相关数据披露。此外,多项dinaciclib与其他药物联用治疗各种肿瘤的临床实验正在进行中。
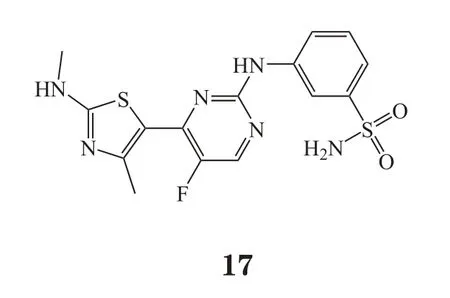
2.1.4 LS-007 LS-007(CDKI-73,17)是笔者所在课题组与澳大利亚南澳大学正在共同研究的新型CDK抑制剂。该化合物是目前已有报道中活性最强的CDK9抑制剂(IC50=4 nmol·L-1)之一,其分子水平也能抑制CDK1、CDK2、CDK7的活性,IC50分别为4、3 和91 nmol·L-1[23]。前期研究表明,LS-007对健康人T细胞和B细胞几乎没有毒性,但对CLL细胞有很好的抑制作用,且与氟达拉滨有很好的协同效果[23]。在卵巢癌中,LS-007通过同时靶向CDK9和eIF4E相关通路来发挥抗肿瘤作用[24]。笔者所在课题组发现,该化合物在淋巴瘤和其他不同类型白血病中也显示了较强的抗肿瘤作用。目前该化合物正处于全面临床前研究阶段。
2.2 CDK选择性抑制剂
2.2.1 Palbociclib CDK抑制剂的研发其实已有很长的历史,但一直未有化合物获得临床认可。2015年2月,美国辉瑞(Pfizer)公司的palbociclib(PD-0332991, 12)获FDA批准与雌激素疗法联合用于雌激素受体阳性乳腺癌患者的治疗,该化合物的成功上市再次掀起了CDK抑制剂的研发热潮。在RB野生型细胞中,palbociclib能抑制CDK4、CDK6的活性(IC50分别为9~11和15 nmol·L-1)从而降低RB磷酸化水平,引起G1期阻滞,抑制多种CDK4扩增肿瘤细胞株(如脂肪肉瘤、MCL、黑色素瘤、乳腺、卵巢和结肠癌)的增殖,在多种异位移植瘤模型、基因工程老鼠模型和人原位移植瘤免疫缺陷动物模型中均有效[25-28]。由于palbociclib的选择性和药动学特性都较好,2004年,此化合物即被作为候选化合物用于治疗MCL、黑色素瘤和胶质瘤[2]。基于其良好的Ⅲ期临床数据,FDA加速批准其与来曲唑联合作为治疗ER+/HER2-绝经期后转移性乳腺癌的一线药物[29]。该药的上市为CDK4/6抑制剂及G1期相关靶点药物的开发提供了强有力的支持。
2.2.2 Ribociclib Ribociclib(LEE011, 9)于2010年首次由美国诺华(Novartis)与Astex联合报道,为又一口服有效的CDK4、CDK6选择性抑制剂(IC50分别为10和39 nmol·L-1),目前正进行Ⅲ期临床研究[2]。与预期一样,ribociclib抑制RB磷酸化,引起G0/G1期阻滞并诱导肿瘤细胞(包括有B-Raf或N-Ras突变的黑色素瘤、乳腺癌、脂肪肉瘤和神经母细胞瘤)衰老[2,30-32]。在临床前体内模型中,ribociclib在CDK4/6或其上游调节信号激活的肿瘤中有效,连续给药可抑制移植瘤的生长,对小鼠体质量则无明显影响。临床实验显示ribociclib对乳腺癌和黑色素瘤有较好药效[33]。与palbociclib一样,中性粒细胞减少症是ribociclib的主要毒性反应[34]。2014年1月,ribociclib进入针对乳腺癌治疗的Ⅲ期临床研究,是目前辉瑞公司产品palbociclib的主要竞争者[35]。
2.2.3 Abemaciclib Abemaciclib(LY2835219,10)属于嘧啶苯并咪唑系列分子,2008年由美国Eli Lilly 开发,也是目前临床进展很好的口服有效的CDK4、CDK6选择性抑制剂(IC50分别为2和5 nmol·L-1),且能通过血脑屏障[36]。在体外激酶实验中发现abemaciclib还能抑制CDK9活性(IC50=59 nmol·L-1)[31]。该化合物体内外均能有效抑制RB磷酸化,引起G0/G1期阻滞。体内靶点抑制研究显示,abemaciclib单剂量作用24 h即诱导完全的周期阻滞并抑制RB-E2F相关调节蛋白的表达[37]。在免疫缺陷小鼠中,连续摄取该化合物56 d,即显示出显著抗肿瘤疗效,且对小鼠体质量无明显影响[38]。由于abemaciclib能有效透过血脑屏障,因此该化合物在动物模型中显著抑制小鼠原位颅内移植脑胶质瘤的生长[37]。与palbociclib和ribociclib不同,abemaciclib可以每天或者每2天给药1次,2天1次给药的最大耐受剂量是200 mg,低于ribociclib,其毒副反应主要包括腹泻、疲劳和中性粒细胞减少症[39]。目前,其正进行针对HER2-的转移性乳腺癌和非小细胞肺癌的Ⅲ期临床研究。
3 非ATP竞争性CDK抑制剂
由于不同CDK之间高度同源,且多数晶体结构未被解析,因此很长时间内,CDK抑制剂并未在临床获得令人满意的结果,研发进展一直比较缓慢。同时,由于CDK在某些细胞中具有特殊功能,因此有些化合物虽然临床前效果显著,但临床应用时因伴随严重毒副作用而使该类药物的开发应用受限(Sausville 等,Trends Mol Med,2002年)。随着研究的深入,人们认为提高CDK抑制剂的选择性可能是其成功开发的关键。过去10多年里,科学家致力于高度选择性CDK抑制剂的开发。由于ATP结合口袋在不同CDK或其他激酶之间相对保守,因此,靶向非ATP结合口袋和干扰蛋白-蛋白结合为开发高选择性CDK抑制剂开辟了新的思路,希望这类抑制剂能避免ATP竞争性抑制剂的诸多副作用[40]。
近来涌现了多种开发CDK/cyclin抑制剂的新方法,包括通过干扰底物识别、靶向必需蛋白-蛋白相互作用、靶向构象改变所必需的残基等[41-42]。此类抑制剂研发策略开始较晚,因此目前该类抑制剂尚较少,主要包括小分子抑制剂和多肽[43]。由于这些小分子抑制剂不与ATP结合位点作用,因此在高浓度的ATP下仍能明显抑制CDK/cyclin复合物的活性。多肽抑制剂则主要为模仿CDK内源抑制子(如p21、p27、p57)的小分子多肽[43-44]。这些小分子多肽为更好地了解新的小分子结合位点和结合方式提供了基础。非ATP竞争性抑制剂主要包括多肽、多肽类似物、合成小分子等,干扰蛋白-蛋白相互作用、作为底 物竞争抑制剂或异构抑制剂等[3,40]。
3.1 底物竞争抑制剂
CDK/cyclin发挥活性过程中,底物与cyclin调节亚基表面疏水的“cyclin groove”结合,聚集并被识别,从而被磷酸化,抑制这一识别可抑制CDK活性[45]。最早开发特异性CDK抑制剂的策略需追溯到1996年,当年有人在研究CDK2/cyclin复合物与周期调节子相互作用时发现,CDK调节子(包括E2F1/2/3、P107、P130、P21、P27、P57)有共同的识别序列——Z-arginine-X-leucine(ZRXL)序列(其中Z和X相对高度保守),含有该序列的多肽能够抑制CDK2/cyclin的活性(Adams等,Mol Cell Biol,1996年)。实验证明,这个cyclin聚集序列(cyclin recruitment motif,CRM)为CDK2-cyclin A/E的底物所共用。这些蛋白包含了一段12个氨基酸的相似区域,负责结合到cyclin结合槽。所有蛋白都需结合到这个裂缝才能被CDK2磷酸化,因此占据cyclin A底物结合槽的分子,能以底物竞争性的方式达到CDK活性的抑制[46]。这个基本的策略随后被REPLACE(replacement with partial ligand alternatives through computational enrichment)拓展,用于开发类药性更好的CDK2多肽类似物抑制剂[47]。
3.2 干扰蛋白-蛋白相互作用的CDK抑制剂
CDK需与其cyclin亚基结合才能发挥活性,因此干扰这一结合可达到抑制CDK活性的目的。此外,不同蛋白(肿瘤抑制子、转录因子等)识别CDK-cyclin通过或至少部分通过蛋白-蛋白相互作用来完成,阻止这些位点可阻止CDK底物的识别和磷酸化[48]。
Cyclin A、cyclin D、cyclin E与CDK的识别位点已被确证,但CDK-cyclin之间的结合很紧密,能有效干扰这两者结合的小分子尚未见报道[42]。然而,多肽类似物可模仿内源性CDK抑制子(p16、p21、p27)或内源性底物(E2F、p53、pRB、p130、p107),干扰CDK与cyclin之间,或CDK/cyclin与p21、p27、p107等之间的结合,也能抑制CDK活性。多肽类似物特异性高,但因其分子大,结构易变,药动学性质差,降解快且难以进入肿瘤细胞等使其在肿瘤病灶处难以达到有效治疗剂量[40,42],影响其进一步研发。然而,最近Wang等[49]发现了一类具有膜通透性的多肽PTD4,能特异性地靶向蛋白-蛋白相互作用界面抑制CDK4/ cyclin D复合物形成,进而体外诱导肿瘤细胞周期阻滞和凋亡,体内显示抗肿瘤效果同时降低副作用。另外,Metamax公司也报道了一类嵌合多肽MM-D37K,可以模仿内源性CDK抑制子p16INK4a的作用,影响CDK活性,发挥抗肿瘤作用[50]。
3.3 异构抑制剂
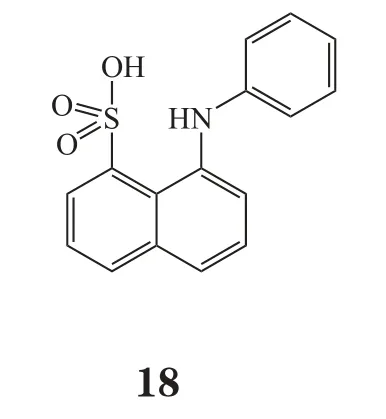
异构抑制剂通常结合到ATP结合口袋附近,但并不占据与ATP结合的序列,其能稳定酶的失活构象,干扰酶活化的构象转变,与底物或ATP竞争[50-51]。由于这类抑制剂针对特定的CDK需要一个特定的环境,因此选择性更好。当前异构抑制剂在ABL/P38和MEK1抑制剂的开发中得到成功应用,因此这是一个充满希望的CDK抑制剂研发策略[52]。最近,Martin等[53]报道了一种用异构作用模式鉴定CDK2小分子抑制剂的新方法,其团队发现了CDK2潜在的异构位点,可以改变CDK2和cyclin A/E的相互作用。此外,该团队还发现2个能够与CDK2相互作用的ANS (8-anilino-1-naphthalene sulfonate,18)分子,一旦ANS分子连接到CDK2上,PSTAIRE 螺旋构象发生改变,使得其不能与cyclin结合[54]。
3.4 共价抑制剂
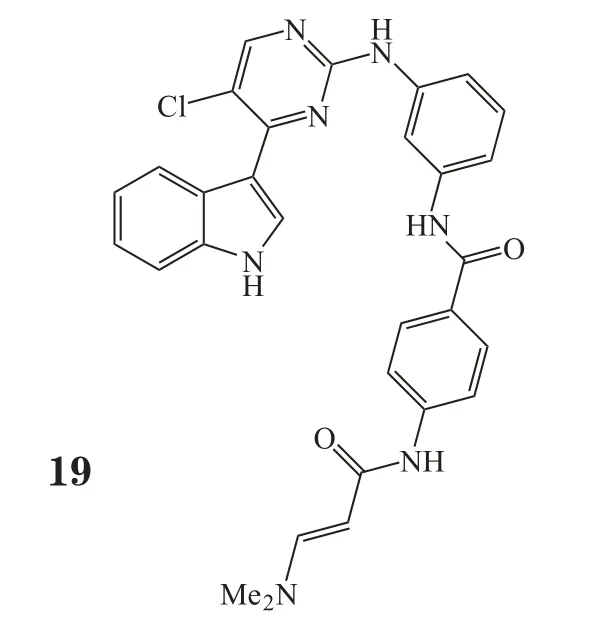
2014年,Gray团队利用能够抑制增殖的表型分析来鉴定激酶的共价抑制剂,发现了CDK7的抑制剂——THZ1(19)。THZ1含有一个丙烯酰胺结构,能共价结合到传统激酶区域外的半胱氨酸残基上,是首个被报道的共价不可逆CDK抑制剂。THZ1在T-ALL和其他造血系统肿瘤中显示了较好疗效[55]。
以上这些工作成果进一步提示非传统的CDK抑制剂(非ATP竞争、异构、共价)在获得CDK家族特异性方面极具潜力。
4 CDK抑制剂与多种药物协同应用于抗肿瘤治疗
许多CDK抑制剂临床前抗肿瘤活性显著,临床效果却与预期不符。相应靶点被抑制后,代偿机制迅速出现,因而即使化合物很好地抑制了靶点活性,却并未抑制肿瘤的发展,且常因伴随有较严重的毒副作用,限制了它们在临床上的应用。
很多临床和临床前实验证实,CDK抑制剂与化疗药物或其他靶向药物之间有很好的协同效果,已上市的palbociclib 也是与雌激素疗法联合用于雌激素受体阳性乳腺癌患者的治疗,这些研究结果提示CDK抑制剂在抗肿瘤的临床治疗上可能更倾向于联合用药从而增强活性,降低毒副作用,进而使患者受益。此外,联合用药能更快、更有效地减少癌变细胞,因而一定程度上能限制耐药的产生。
研究表明,CDK抑制剂与很多抗肿瘤药物有协同增效的作用。CDK抑制剂与细胞毒类药物[如5-FU、顺铂、多柔比星(doxorubicin)、紫杉醇(taxol)等]协同明显。Flavopiridol与阿糖胞苷(cytarabine)和米托蒽醌(mitoxantrone)联用治疗初发AML的Ⅱ期临床研究中,有约70%的病人有完全反应[56]。当化疗药物先于CDK抑制剂给药,细胞先同步或阻滞在某一周期时,CDK抑制剂活性更强。如将阿糖胞苷、紫杉醇或多柔比星等先作用24 h,使细胞阻滞在G1或G2/M期后再给flavopiridol作用,比同时给药效果更好[57]。同样,许多化疗药物在S或G2/M期发挥药效,若用CDK1/2抑制剂(如dinacicilib)将细胞阻滞在这一期后再配合使用相应的化疗药物则可以产生更强的治疗作用。此外,抑制某些CDK的活性可通过降低抗凋亡蛋白的表达协同增强化疗药物药效。如dinacicilib通过抑制CDK7/9下调抗凋亡蛋白(如Mcl-1等)表达,使原代CLL细胞对长春新碱(vincristine)更敏感[58]。
虽然CDK4/6抑制剂可将肿瘤细胞阻滞在G0/G1期,使得细胞对在S或G2/M期发挥药效的化疗药物不敏感,但它们能增强其他一些靶向药物的药效。临床前数据表明palbociclib与蛋白酶体抑制剂硼替佐米(bortezomib)和地塞米松(dexamethasone)共同处理可增加MM细胞的死亡,这一现象在临床病人中也得到证实。Ⅰ期临床研究显示,在这种联合方案中,硼替佐米以低于其他联合研究时采用的剂量仍能产生药效;Ⅱ期临床研究显示,该联合给药治疗在20%的病人中产生疗效[59-60]。在K-Ras突变的结肠癌人源异种移植(patient-derived xenograft,PDX)模型中,palbociclib与MEK抑制剂也体现出较好的协同作用[61]。在胰腺癌患者中,CDK4/6抑制剂与IGF1R抑制剂和mTOR抑制剂也表现出协同作用[62-63]。此外,CDK4/6抑制剂与多种激素疗法协同效应显著。在针对ER+乳腺癌的Ⅱ期临床研究中,palbociclib与芳香化酶抑制剂来曲唑有很好的协同作用,联用组病情缓解率达52%,而来曲唑单药治疗组缓解率仅32%[64]。与来曲唑单药治疗相比,该联合疗法显著延长了ER+/HER2-绝经期后乳腺癌患者的无进展生存期(10.2个月vs 20.2个月)[65]。基于此结果,2015年2月,美国FDA加速批准了palbociclib与来曲唑联合治疗ER+/HER2-绝经期后乳腺癌,palbociclib成为首个上市的CDK抑制剂,这是该领域数十年研发历史的重大突破。
此外,CDK抑制剂也能够逆转化疗和放疗中出现的耐药现象。CDK抑制剂SNS-032单药使用对原代AML细胞有明显的增殖抑制活性,与阿糖胞苷还有显著协同作用,更有趣的是,该化合物还可使已对化疗耐受的NSCLC细胞对放疗更加敏感[66]。已明确MAPK活化及cyclin D表达增加可介导B-Raf抑制剂威罗非尼(vemurafenib)的耐药,而abemaciclib则能克服这些耐药因素,增强威罗非尼抗肿瘤活性并延迟肿瘤复发[67]。
5 展望
CDK抑制剂虽然已成功用于肿瘤临床治疗中,但其在基础研究中仍有很多未知,使该类抑制剂的疗效、适应证、敏感人群的选择等仍不确定。
与第1代CDK抑制剂相比,第2代CDK抑制剂,特别是在临床已获成功的palbociclib等,选择性更好,治疗指数更高,毒副作用更小,提示提高CDK抑制剂的选择性对开发出成功的CDK抑制剂很关键。为此,在研发策略上,研究人员也已从寻找发现ATP竞争性CDKs抑制剂的单一手段中走出,着手发现非ATP竞争、异构、共价等不同作用方式的非“传统”的CDK抑制剂[48],从而使研发选择性更好的CDK抑制剂成为可能。但是尽管人们当前更致力于开发单一CDK选择性的抑制剂,其实尚不能完全确定这就是CDK抑制剂研发的最佳选择。CDK家族成员众多,它们在不同肿瘤中的表达也有差异,在特定肿瘤中,哪种CDK活性失调、在该肿瘤发生发展中作用更显著尚不明确,此外各个CDK/cyclin复合物的具体生理功能也并非全面解析,这些基础研究的短板阻碍了CDK抑制剂的进一步发展。
另外,在CDK抑制剂临床应用中,寻找敏感生物标志物、挑选合适病人显得尤为重要。缺乏好的生物标志物,无法筛选合适基因背景的肿瘤病人,不能准确预测肿瘤对CDK抑制剂的反应是导致临床上众多CDK抑制剂失败的原因之一[22]。Flavopiridol和dinaciclib都在CLL病人中效果很好且少有例外,表明确实存在对这类化合物敏感的分子基础。CDK4抑制剂单独作用能够抑制RAS或HER2诱导肿瘤的生长,对Myc诱导的肿瘤无效,而CDK1抑制对Myc诱发的淋巴瘤和肝母细胞瘤有效[68-70]。这些研究结果提示,生物标志物和基因背景的鉴定对CDK抑制剂临床应用有很重要的价值。
肿瘤是一个异质性非常高的疾病,就目前的临床结果而言,CDK抑制剂联合用药比单药使用在肿瘤的临床治疗中更有前景。因此,应该进一步探索CDK抑制剂与其他抗肿瘤药物联合用药的可能性,找到合理有效的联合用药策略,加速推动CDK抑制剂在临床应用上的进展。
[1]Lim S, Kaldis P. CDKs cyclins and CKIs: Roles beyond cell cycle regulation [J]. Development, 2013, 140(15): 3079-3093.
[2]Asghar U, Witkiewicz A K, Turner N C, et al. The history and future of targeting cyclin-dependent kinases in cancer therapy [J]. Nat Rev Drug Discov, 2015, 14(2): 130-146.
[3]Cicenas J, Kalyan K, Sorokinas A, et al. Highlights of the latest advances in research on CDK inhibitors [J]. Cancers, 2014, 6(4): 2224-2242.
[4]Canavese M, Santo L, Raje N. Cyclin dependent kinases in cancer [J]. Cancer Biol Ther, 2014, 13(7): 451-457.
[5]Okamoto M, Hidaka A, Toyama M, et al. Selective inhibition of HIV-1 replication by the CDK9 inhibitor FIT-039 [J]. Antiviral Res, 2015, 123: 1-4.
[6]Guen V J, Gamble C, Flajolet M, et al. CDK10/cyclin M is a protein kinase that controls ETS2 degradation and is deficient in STAR syndrome [J]. Proc Natl Acad Sci USA, 2013, 110(48): 19525-19530.
[7]Malumbres M. Cyclin-dependent kinases [J]. Genome Biol, 2014, 15(6): 122.
[8]Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: A changing paradigm [J]. Nat Rev Cancer, 2009, 9(3): 153-166.
[9]Peyressatre M, Prevel C, Pellerano M, et al. Targeting cyclin-dependent kinases in human cancers: From small molecules to peptide inhibitors [J]. Cancers:Basel, 2015, 7(1): 179-237.
[10]Mariaule G, Belmont P. Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where are we now? A short survey [J]. Molecules,2014, 19(9): 14366-14382.
[11]Bose P, Simmons G L, Grant S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies [J]. Expert Opin Investig Drugs,2013, 22(6): 723-738.
[12]Phelps M A, Lin T S, Johnson A J, et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia [J]. Blood, 2009,113(12): 2637-2645.
[13]Lin T S, Ruppert A S, Johnson A J, et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease [J]. J Clin Oncol, 2009, 27(35): 6012-6018.
[14]Christian B A, Grever M R, Byrd J C, et al. Flavopiridol in chronic lymphocytic leukemia: A concise review [J]. Clin Lymphoma Myeloma,2009, 9 Suppl 3: S179-S185.
[15]Wang L M, Ren D M. Flavopiridol, the first cyclin-dependent kinase inhibitor: Recent advances in combination chemotherapy [J]. Mini Rev Med Chem, 2010, 10(11): 1058-70.
[16]Berberich N, Uhl B, Joore J, et al. Roscovitine blocks leukocyte extravasation by inhibition of cyclin-dependent kinases 5 and 9 [J]. Br J Pharmacol, 2011, 163(5): 1086-98.
[17]MacCallum D E, Melville J, Frame S, et al. Seliciclib (CYC202,R-roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1 [J]. Cancer Res, 2005, 65(12): 5399-5407.
[18]Cicenas J, Kalyan K, Sorokinas A, et al. Roscovitine in cancer and other diseases [J]. Ann Transl Med, 2015, 3(10): 135.
[19]Parry D, Guzi T, Shanahan F, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor [J]. Mol Cancer Ther,2010, 9(8): 2344-2353.
[20]Nemunaitis J J, Small K A, Kirschmeier P, et al. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies[J]. J Transl Med, 2013, 11:259.
[21]Kumar S K, LaPlant B, Chng W J, et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma [J]. Blood, 2015, 125(3): 443-448.
[22]Guha M. Cyclin-dependent kinase inhibitors move into phase III [J]. Nat Rev Drug Discov, 2012, 11(12): 892-894.
[23]Walsby E, Pratt G, Shao H, et al. A novel CDK9 inhibitor preferentially targets tumor cells and synergizes with fludarabine [J]. Oncotarget,2014, 5(2): 375-385.
[24]Lam F, Abbas A Y, Shao H, et al. Targeting RNA transcription and translation in ovarian cancer cells with pharmacological inhibitor CDKI-73 [J]. Oncotarget, 2014, 5(17): 7691-7704.
[25]Young R J, Waldeck K, Martin C, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines[J]. Pigment Cell Melanoma Res, 2014, 27(4): 590-600.
[26]Leonard J P, LaCasce A S, Smith M R, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma [J]. Blood, 2012, 119(20): 4597-4607.
[27]Barton K L, Misuraca K, Cordero F, et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma [J]. PLoS One, 2013, 8(10): e77639.
[28]Rihani A, Vandesompele J, Speleman F, et al. Inhibition of CDK4/6 as a novel therapeutic option for neuroblastoma [J]. Cancer Cell Int, 2015,15 :76.
[29]VanArsdale T, Boshoff C, Arndt K T, et al. Molecular pathways: Targeting the cyclin D-CDK4/6 axis for cancer treatment [J]. Clin Cancer Res, 2015, 21(13): 2905-2910.
[30]Rader J, Russell M R, Hart L S, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma [J]. Clin Cancer Res, 2013, 19(22): 6173-6182.
[31]Dickson M A. Molecular pathways: CDK4 inhibitors for cancer therapy[J]. Clin Cancer Res, 2014, 20(13): 3379-3383.
[32]Drug combo shows promise in NRAS-mutant melanoma [J]. Cancer Discov, 2014, 4(8): OF2.
[33]Infante J R, Shapiro G I, Witteveen P O, et al. Abstract a276: Phase 1 multicenter, open label, dose-escalation study of LEE011, an oral inhibitor of cyclin-dependent kinase 4/6, in patients with advanced solid tumors or lymphomas [J]. Mol Cancer Therap, 2013, 12(11 Suppl): A276.
[34]Mayer E L. Targeting breast cancer with CDK inhibitors [J]. Curr Oncol Rep, 2015, 17(5): 443.
[35]Dukelow T, Kishan D, Khasraw M, et al. CDK4/6 inhibitors in breast cancer [J]. Anticancer Drugs, 2015, 26(8): 797-806.
[36]Raub T J, Wishart G N, Kulanthaivel P, et al. Brain exposure of two selective dual CDK4 and CDK6 inhibitors and the antitumor activity of CDK4 and CDK6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft [J]. Drug Metab Dispos, 2015,43(9): 1360-1371.
[37]Gelbert L M, Cai S, Lin X, et al. Abstract b233: Identification and characterization of LY2835219: A potent oral inhibitor of the cyclindependent kinases 4 and 6 (CDK4/6) with broad in vivo antitumor activity [J]. Mol Cancer Ther, 2011, 10(11 Suppl): B233.
[38]Gelbert L M, Cai S, Lin X, et al. Preclinical characterization of the cdk4/6 inhibitor ly2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine [J]. Invest New Drugs, 2014, 32(5): 825-837.
[39]Patnaik A, Rosen L S, Tolaney S M, et al. Abstract ct232: Clinical activity of LY2835219, a novel cell cycle inhibitor selective for CDK4 and CDK6, in patients with metastatic breast cancer [J]. Cancer Res,2014, 74(19 Suppl): CT232.
[40]Peyressatre M, Prével C, Pellerano M, et al. Targeting cyclin-dependent kinases in human cancers: From small molecules to peptide inhibitors [J].Cancers, 2015, 7(1): 179-237.
[41]Cohen P, Alessi D R. Kinase drug discovery--- what's next in the field? [J]. ACS Chem Biol, 2013, 8(1): 96-104.
[42]Fang Z, Grutter C, Rauh D. Strategies for the selective regulation of kinases with allosteric modulators: Exploiting exclusive structural features [J]. ACS Chem Biol, 2013, 8(1): 58-70.
[43]Abate A A, Pentimalli F, Esposito L, et al. ATP-noncompetitive CDK inhibitors for cancer therapy: An overview [J]. Exp Opin Investig Drugs,2013, 22(7): 895-906.
[44]Cirillo D, Pentimalli F, Giordano A. Peptides or small molecules?Different approaches to develop more effective CDK inhibitors [J]. Curr Med Chem, 2011, 18(19): 2854-2866.
[45]Harrison S. Non-ATP-competitive kinase inhibitors-enhancing selectivity through new inhibition strategies [J]. Expert Opin Drug Discov , 2008, 3(7): 761-774
[46]Canela N, Orzaez M, Fucho R, et al. Identification of an hexapeptide that binds to a surface pocket in cyclin A and inhibits the catalytic activity of the complex cyclin-dependent kinase 2-cyclin A [J]. J Biol Chem, 2006, 281(47): 35942-35953.
[47]Liu S, Premnath P N, Bolger J K, et al. Optimization of non-ATP competitive CDK/cyclin groove inhibitors through replace-mediated fragment assembly [J]. J Med Chem, 2013, 56(4): 1573-1582.
[48]Mariaule G, Belmont P. Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where are we now? A short survey [J]. Molecules,2014, 19(9): 14366-14382.
[49]Wang H, Chen X, Chen Y, et al. Antitumor activity of novel chimeric peptides derived from cyclind/CDK4 and the protein transduction domain 4 [J]. Amino Acids, 2013, 44(2): 499-510.
[50]Vladimir K B, Tatyana M K, Elena A K, et al. New targeted anti CDK4/6 peptide MM-D37K[J].J Clin Oncol, 2013, 31(suppl15): e13545.
[51]Hu Y, Li S, Liu F, et al. Discovery of novel nonpeptide allosteric inhibitors interrupting the interaction of CDK2/cyclin A3 by virtual screening and bioassays [J]. Bioorg Med Chem Lett, 2015, 25(19): 4069-4073.
[52]Eglen R M, Reisine T. Human kinome drug discovery and the emerging importance of atypical allosteric inhibitors [J]. Expert Opin Drug Discov, 2010, 5(3): 277-290.
[53]Martin M P, Alam R, Betzi S, et al. A novel approach to the discovery of small-molecule ligands of CDK2 [J]. Chembiochem, 2012, 13(14): 2128-2136.
[54]Betzi S, Alam R, Martin M, et al. Discovery of a potential allosteric ligand binding site in CDK2 [J]. ACS Chem Biol, 2011, 6(5): 492-501.
[55]Kwiatkowski N, Zhang T, Rahl P B, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor [J]. Nature, 2014,511(7511): 616-620.
[56]Zeidner J F, Foster M C, Blackford A L, et al. Randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine, and mitoxantrone(FLAM) versus cytarabine/daunorubicin (7+3) in newly diagnosed acute myeloid leukemia [J]. Haematologica, 2015, 100(9): 1172-1179.
[57]Song Y, Xin X, Zhai X, et al. Sequential combination of flavopiridol with taxol synergistically suppresses human ovarian carcinoma growth[J]. Arch Gynecol Obstet, 2015, 291(1): 143-150.
[58]Bates D J, Salerni B L, Lowrey C H, et al. Vinblastine sensitizes leukemia cells to cyclin-dependent kinase inhibitors, inducing acute cell cycle phase-independent apoptosis [J]. Cancer Biol Ther, 2011, 12(4): 314-325.
[59]Niesvizky R, Badros A Z, Costa L J, et al. Phase 1/2 study of cyclindependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma[J]. Leuk Lymphoma, 2015, 56(12):3320-3328.
[60]Altenburg J D, Farag S S. The potential role of PD0332991 (palbociclib)in the treatment of multiple myeloma [J]. Expert Opin Investig Drugs,2015, 24(2): 261-271.
[61]Li J, Xu M, Yang Z, et al. Simultaneous inhibition of MEK and CDK4 leads to potent apoptosis in human melanoma cells [J]. Cancer Invest,2010, 28(4): 350-356.
[62]Franco J, Witkiewicz A K, Knudsen E S. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer [J]. Oncotarget, 2014, 5(15): 6512-6525.
[63]Heilmann A M, Perera R M, Ecker V, et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16Ink4A-deficient pancreatic cancers [J]. Cancer Res, 2014, 74(14): 3947-3958.
[64]Finn R S, Crown J P, Lang I, et al. The cyclin-dependent kinase 4/6inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative,advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study [J]. Lancet Oncol, 2015, 16(1): 25-35.
[65]Dhillon S. Palbociclib: First global approval [J]. Drugs, 2015, 75(5): 543-551.
[66]Walsby E, Lazenby M, Pepper C, et al. The cyclin-dependent kinase inhibitor SNS-032 has single agent activity in AML cells and is highly synergistic with cytarabine [J]. Leukemia, 2011, 25(3): 411-419.
[67]Yadav V, Burke T F, Huber L, et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation [J]. Mol Cancer Ther, 2014, 13(10): 2253-2263.
[68]Mao C Q, Xiong M H, Liu Y, et al. Synthetic lethal therapy for KRAS mutant non-small-cell lung carcinoma with nanoparticle-mediated CDK4 siRNA delivery [J]. Mol Ther, 2014, 22(5): 964-973.
[69]Witkiewicz A K, Cox D, Knudsen E S. CDK4/6 inhibition provides a potent adjunct to HER2-targeted therapies in preclinical breast cancer models [J]. Genes Cancer, 2014, 5(7/8): 261-272.
[70]Kang J, Sergio C M, Sutherland R L, et al. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells [J]. BMC Cancer, 2014,14(32): 1-13.

[专家介绍] 陈奕 :2004年毕业于中国科学院上海药物研究所,获博士学位。现为中国科学院上海药物研究所研究员,博士生导师,中国抗癌协会药物专业委员会和中国药理学会肿瘤药理专业委员会委员。主要从事抗肿瘤药物靶标发现、新药研发及作用机制确证。已在Clin Cancer Res、Oncotarget、J Natl Cancer Inst等杂志发表SCI论文50余篇,获授权国际国内专利9项;迄今主持包括科技部重大专项、国家自然科学基金、国家重大研究计划等多个项目。
Progress in Development of Cyclin-dependent Kinase Inhibitors as Cancer Therapy
XIE Shao, DING Jian, CHEN Yi
(Division of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica,Chinese Academy of Sciences, Shanghai 201203, China)
Cyclin-dependent kinases(CDKs) play critical roles in controlling cell cycle and regulating cell proliferation, transcription and survival. CDKs are often abnormally activated in numerous tumors and thus become attractive anticancer targets. Palbociclib, a CDK4/CDK6 inhibitor, was approved by the US Food and Drugs Administration in 2015, Dozens of compounds targeting CDKs are currently in clinical development for various solid tumors and hematopoietic malignances.In this article, the recent studies on CDK inhibitors, related challenges and solutions were reviewed, and the probable clinical application of CDK inhibitors was also discussed.
CDK; inhibitor; ATP competitive; non-ATP competitive; combined therapy
R966;R962
A
1001-5094(2015)10-0734-12
接受日期:2015-10-08
*通讯作者:陈奕,研究员;
研究方向:肿瘤药理学;
Tel:021-50806600-4306; E-mail:ychen@simm.ac.cn
