CRISPR/Cas9系统介导小鼠MSTN基因定点修饰研究
2015-11-12任红艳毕延震李莉等
任红艳 毕延震 李莉 等

摘要:利用CRISPR/Cas9系统突变小鼠Myostatin基因,为制备MSTN基因修饰猪奠定了理论和实践基础。结果表明,成功构建了CRISPR/Cas9定点敲除技术平台,并获得MSTN基因发生缺失突变的小鼠模型。
关键词:CRISPR/Cas9系统;Myostain基因;定点修饰
中图分类号:Q78 文献标识码:A 文章编号:0439-8114(2015)20-5155-04
DOI:10.14088/j.cnki.issn0439-8114.2015.20.058
Production of MSTN Gene Mutant Mice Using CRISPR/Cas9 System
REN Hong-yan,BI Yan-zhen, LI Li, ZHENG Xin-min
(Institute of Animal Husbandry and Veterinary, Hubei Academy of Agricultural Sciences, Wuhan 430064, China)
Abstract: In this paper, Myostatin gene mutant mice were generated using CRISPR/Cas9 system, which can be further applied to preparation of transgenic pigs. The results showed that CRISPR/Cas9 platform was established and MSTN gene mutant mice were generated successfully.
Key words:CRISPR/Cas9 system; Myostatin gene; site-directed mutation
基因敲除动物模型是开展基因功能研究、寻找合适药物作用靶点的重要工具。传统的基因敲除方法需要通过复杂的打靶载体构建、ES细胞筛选、嵌合体选育等步骤,不仅流程繁琐,对技术的要求很高,而且费用大、耗时较长、成功率也较低。即使对于技术比较成熟的实验室,利用传统技术构建基因敲除大、小鼠一般也需要一年以上时间,因此其应用受到限制。新一代人工核酸内切酶(Engineered endonuclease,EEN)的出现,使这一现状彻底改观,锌指核酸内切酶(Zinc Finger Endonuclease,ZFN)是第一代人工核酸内切酶,其可以在各种复杂基因组的特定位置制造DNA的双链切口[1]。截至目前,ZFN已经成功应用于黑长尾猴、大鼠、小鼠、中国仓鼠、斑马鱼、果蝇、海胆、家蚕、拟南芥、烟草、玉米、猪、牛、人类iPS细胞中[2]。第二代人工核酸酶是一类转录激活因子效应物核酸酶(Transcription activator-like effector nuclease,TALEN),可以和ZFN一样对复杂的基因组进行精细的修饰,同时其构建较为简单,特异性更高,因此受到了科研工作者的青睐,在植物、人类细胞、小鼠、斑马鱼、猪、牛中迅速得以应用[3-5]。
2013年,《Cell》杂志报道称加州大学旧金山分校的研究人员发现了一种效率更高、修饰更为精确的敲除基因的方法,称为CRISPR/Cas9系统[6],该系统因其操作简单、成本低、效率高等特点受到国内外研究者的关注。CRISPR/Cas9是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御,可用来对抗入侵的病毒及外源DNA[7]。CRISPR/Cas9系统通过将入侵噬菌体和质粒DNA的片段整合到CRISPR中,并利用相应的CRISPR RNAs(crRNAs)来指导同源序列的降解,从而提供免疫性[8]。该系统的工作原理是crRNA(CRISPR-derived RNA)通过碱基配对与tracrRNA(trans-activating RNA)结合形成 tracrRNA/crRNA 复合物,此复合物引导核酸酶Cas9蛋白质在与crRNA配对的序列靶位点剪切双链DNA。而通过人工设计的这两种RNA,可以改造形成具有引导作用的sgRNA(short guide RNA),从而引导Cas9对DNA的定点修饰。2013年,美国两个实验室报道了基于CRISPR/Cas9系统在细胞系中进行基因敲除的新方法,利用靶点特异性的RNA将Cas9核酸酶带到基因组上的具体靶点,从而对特定基因位点进行切割导致突变,并迅速将该技术应用于基因敲除小鼠和大鼠的制备中[8,9]。Jaenisch等利用CRISPR/Cas9系统在3~4周时间内构建出携带5种突变的小鼠,而用传统的方法则需要3~4年的时间[10]。CRISPR/Cas9技术成为继锌指核酸酶(ZFN)、ES 细胞打靶和 TALEN 等技术后可构建基因敲除动物的第四种方法,且有效率高、速度快、生殖系转移能力强等特点,在动物模型的构建中应用前景非常广阔。
肌肉生长抑制素(Myostatin,MSTN),又称生长分化因子-8,属于转化生长因子-β超家族,是骨骼肌发育的主要负调控因子。MSTN基因通过抑制肌源性调控因子如MyoD等降低成肌细胞的增殖率,并抑制肌细胞分化,MSTN基因缺失的动物表现明显的双肌表型[11]。因此,敲除MSTN基因成为提高动物瘦肉率的重要手段。本研究建立了CRISPR/Cas9系统介导的基因定点修饰技术平台,并应用该技术制备了MSTN基因突变的小鼠,为后期制备转基因猪奠定了理论和实践基础。
1 材料与方法
1.1 细菌与质粒
限制性内切酶、dNTP Mix、高保真酶rTaq、pMD-18T载体和T4 DNA连接酶,购自宝生物工程(大连)有限公司;质粒DNA小量提取试剂盒,琼脂糖凝胶DNA回收试剂盒购自天根生化科技(北京)有限公司;大肠杆菌DH5α为湖北省农业科学院畜牧兽医研究所生物技术室存留;MLM3613和pDR274载体购自美国Addgene公司;体外转录试剂盒mMessagemMachine T7购自Ambion公司,SURVEYOR突变检测试剂盒购自Transgenomic公司。
1.2 靶位点设计
从NCBI(http://archive-dtd.ncbi.nlm.nih.gov/)获得小鼠MSTN基因组序列,利用ZiFit软件预测靶位点,选取其中的3个位点。
1.3 Cas9 mRNA的体外转录和加尾
1.3.1 Cas9表达载体线性化 Cas9表达载体MLM3613提纯后用PmeⅠ酶切线性化。利用PCR产物纯化试剂盒回收线性化片段;向回收产物中加入2倍体积无水乙醇和1/10体积NaAc,-20 ℃放置过夜后高速离心回收产物,无RNA酶水溶解产物浓度至500~1 000 ng/μL,-20 ℃保存备用。
1.3.2 体外转录 利用Ambion公司mMessagemMachine T7 试剂盒进行Cas9 mRNA的体外转录和polyA加尾。20 μL体外转录体系为:2 μL线性化质粒DNA(1 μg),10 μL 2×NTP/CAP, 2 μL 10×Reaction Buffer,2 μL Enzyme mix,4 μL ddH2O,混匀后37 ℃反应2 h。80 μL polyA加尾体系为:20 μL 5×EPAP Buffer,10 μL MgCl2(25 mmol/L),10 μL ATP(10 mmol/L),4 μL E-PAP,36 μL ddH2O,混匀后37 ℃反应30 min。以1%琼脂糖凝胶检测RNA转录和加尾情况。
1.4 gRNA编码载体构建及体外转录
根据软件预测的3个靶序列分别合成序列互补的正、反向引物,在正向引物5端合成TAGG接头,在反向引物5端合成CAAA接头,设计模式如图1所示,合成3个靶序列引物如表1所示。
gRNA体外转录载体pDR274载体购自Addgene公司(Addgene No. 42250),将该载体用Bsa Ⅰ酶切线性化后回收,与上面得到的双链产物进行连接,连接产物转化大肠杆菌DH5α后提取质粒进行鉴定。将阳性质粒大量提取后用Dra Ⅰ内切酶线性化并回收线性化产物,浓缩浓度至500~1 000 ng/μL,-20 ℃保存备用。
1.5 小鼠胚胎注射
选取4~5周龄母鼠作为受精卵供体,腹腔注射PMSG和hCG进行超数排卵,交配后取细胞期胚胎进行显微注射。将Cas9 mRNA(100 ng/μL)和sgRNA(100 ng/μL)混合均匀,注射到细胞期胚胎的胞质内,注射后的胚胎移植到假孕母鼠的输卵管内。
1.6 敲除小鼠检测
待移植后的胚胎发育到13 d时,取出胚胎提取基因组DNA进行错配内切酶鉴定。具体步骤为:提取基因组DNA,设计引物扩增出靶序列两侧共350 bp片段,纯化PCR产物;将纯化的PCR产物在95 ℃变性10 min,之后缓慢降温至室温使片段重新退火,降温程序为95℃→85 ℃(每秒降温2 ℃),85 ℃→25 ℃(每秒降温0.5 ℃);重新退火的PCR产物用2 U的T7EI在37 ℃酶切3 h,然后1%琼脂糖凝胶电泳检测结果。
2 结果与分析
2.1 Cas9表达载体构建与线性化
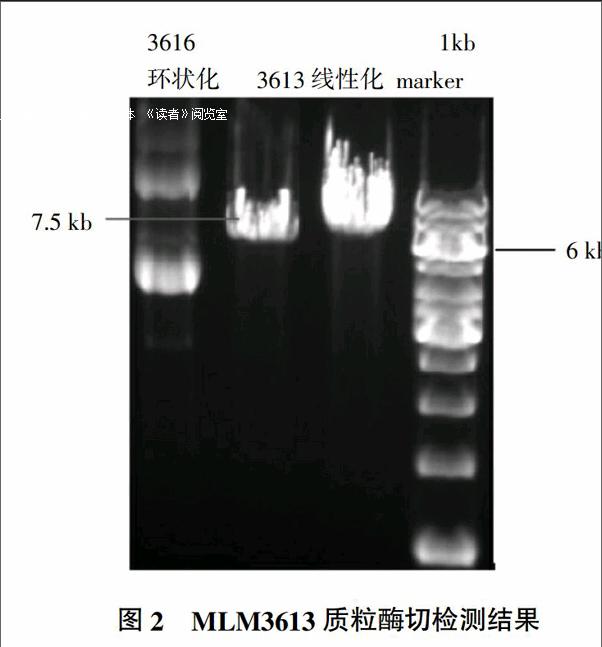
Cas9表达载体MLM3613质粒经PmeⅠ酶切得到7.5 kb线性化片段(图2),乙醇沉淀浓度达到1 000 ng/μL,纯度高,可以用于下一步体外转录试验。
2.2 Cas9 mRNA体外转录结果
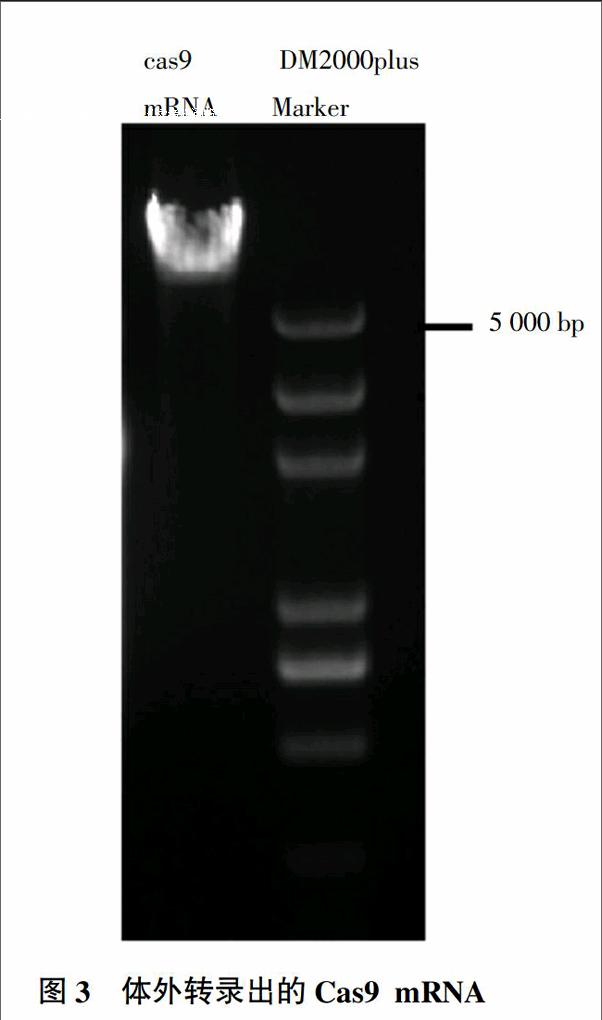
Cas9表达载体经酶切线性化后,通过ABI公司的体外转录试剂盒进行转录,得到了带有帽子结构的Cas9 mRNA,并通过酚仿抽提得到纯化的mRNA。结果(图3)表明,转录的mRNA呈现明显的两条带,为RNA的不同结构,纯度较高,可用于下一步加尾试验。
2.3 加polyA尾后得到全长mRNA序列
将体外转录出的mRNA通过polyA Tailing Kit试剂盒进行加尾,加上长度约150 bp的polyA尾巴,使之成为完整的有功能的mRNA。结果(图4)表明,加尾后的mRNA在电泳时出现了适当的滞后,polyA加尾成功。
2.4 敲除小鼠检测
待孕母鼠怀孕13 d时取出30只胚胎,提取其基因组DNA。用Transgenomic公司的SURVEYOR突变检测试剂盒进行突变检测。结果表明,PCR扩增出T1靶位点两侧473 bp序列,靶序列两侧序列分别为322 bp和151 bp。当序列内部存在碱基突变时,退火后形成的杂合双链能够被酶切开,形成3条带(图5样品10);当不存在碱基突变时,退火形成的双链不存在碱基错配,不能被酶切开,30个样品中共检测出2只为突变阳性。
3 小结与讨论
本研究获得的Cas9和gRNA表达载体,为下一步进行相关研究提供了试验材料;建立了CRISPR/Cas9基因定点修饰技术平台,为转基因小鼠和转基因猪的制备奠定了技术基础,通过CRISPR/Cas9系统获得了MSTN基因敲除小鼠。
基因敲除动物模型一直是在活体动物上开展基因功能研究、寻找合适药物作用靶标的重要工具。但是传统的基因敲除方法需要通过复杂的打靶载体构建、ES细胞筛选、嵌合体小鼠选育等一系列步骤,不仅流程繁琐、技术要求高,而且费用大、耗时较长,成功率受到多方面因素的限制。即使对于技术比较成熟的实验室,利用传统技术构建基因敲除大、小鼠一般也需要一年以上时间。本研究通过体外转录和显微注射等方法在较短时间内获得了MSTN基因敲除的小鼠,从时间、成本等方面节约了大量资源,也为下一步进行大动物转基因研究奠定了基础。但是从检测的结果来看,效率并不高,30只小鼠中只检测到2只阳性。分析原因可能是:①RNA显微注射过程中RNA降解问题。RNA很不稳定,容易受到外界环境中RNA酶的降解而失效,在进行中RNA裸露在环境中的时间较长,且温度无法控制到很低,导致RNA降解严重;②检测方法灵敏度较低。本研究应用的是SURVEYOR检测方法,该方法对试验条件要求特别严格,酶添加量及酶切时间长短都会严重影响试验结果,导致试验结果不稳定,重复性差,只能通过大量克隆测序来解决。
参考文献:
[1] KIM Y G, CHA J, CHANDRASEGARAN S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain[J]. Proceedings of the National Academy of Sciences of the United States of America,1996, 93(3):1156-1160.
[2] URNOV F D, REBAR E J, HOLMES M C,et al. Genome editing with engineered zinc finger nucleases[J]. Nature Reviews Genetics, 2010, 11(9):636-646.
[3] MOSCOU M J, BOGDANOVE A J. A simple cipher governs DNA recognition by TAL effectors[J]. Science, 2009, 326:1501-1508.
[4] BOCH J, SCHOLZE H, SCHORNACK S,et al. Breaking the code of DNA binding specificity of TAL-type III effectors[J]. Science, 2009, 326:1509-1512.
[5] BOCH J, BONAS U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function[J]. Annual review of phytopathology, 2010, 48:419-436.
[6] LI T, HUANG S, ZHAO X, et al. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes[J]. Nucleic Acids Research, 2011, 39(14):6315-6325.
[7] WIEDENHEFT, STERNBERG B S H, DOUDNA J A, et al. RNA-guided genetic silencing systems in bacteria and archaea[J]. Nature,2012, 482: 331-338.
[8] MALI P, ESVELT K M, CHURCH G M, et al. Cas9 as a versatile tool for engineering biology[J]. Nat Methods,2013, 10(10): 957-963.
[9] MALI P, YANG L, ESVELT K M, et al. RNA-guided human genome engineering via Cas9[J]. Science,2013, 339:823-826.
[10] WANG H Y, YANG H, SHIVALILA C S,et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J]. Cell,2013, 153(4): 910-918.
[11] STINCKENS A, LUYTEN T,BIJTTEBIER J, et al.Characterization of the complete porcine MSTN gene and expression levels in pig breeds differing in muscularity[J]. Anim Genet, 2008,39(6):586-596.
