坎地沙坦酯中的潜在基因毒性杂质控制浅析
2015-10-13钱志英赵斌锋
钱志英,赵斌锋
坎地沙坦酯中的潜在基因毒性杂质控制浅析
钱志英,赵斌锋
(浙江金立源药业有限公司,浙江绍兴312369)
以警示结构为判断依据,首先分析了原料药坎地沙坦酯的合成工艺中的潜在基因毒性杂质,然后建立了这类杂质的控制限度要求,最后通过举例其中的溴氰基联苯杂质的控制来阐述这类杂质的控制方法。
坎地沙坦酯;基因毒性;杂质控制
基因毒性化合物是指能直接或间接损伤DNA,使其产生突变和致癌作用的化合物。这类化合物作为非药用成分存在于原料药中则成为基因毒性杂质。在药物合成、纯化和储存运输等过程中,都可能产生基因毒性杂质。因此在药品的研发过程中,对其进行讨论和分析,这也是欧美国家近年来新提出的一个要求。潜在基因毒性杂质是指显示有基因毒性警示结构但未经过实验检测模型检测的杂质,此处的“潜在”与基因毒性相关,与该杂质的存在与否无关。对于潜在基因毒性杂质控制,FDA和EMA等机构在近几年陆续出台了一些指南和技术解答资料,但是目前在国内还没有相关的规定和指南,因此对国内很多人来说,如何判断是否有潜在基因毒性,如何对其残留限度进行设定,设定限度后如何进行监测等等都存在很多疑惑。本文通过解析原料药坎地沙坦酯的合成工艺中可能存在的潜在基因毒性杂质,以期到达探讨这一类杂质的控制方法的目的。
1 坎地沙坦酯的合成路线
坎地沙坦酯为一前体药物,其在体内吸收时被水解成活性代谢物坎地沙坦;坎地沙坦是一种具有高度选择性长效AT1受体拮抗剂(AT1RA),属血管紧张素Ⅱ(AngⅡ)受体抑制剂。与同类产品相,比坎地沙坦酯具有临床疗效优势、良好耐受性和优越的合并用药等特点。
目前,国内外报道制备坎地沙坦酯方法的文献很多,本文选取其中之一[1]进行解析和阐述:工艺过程如下:以3-硝基邻苯二甲酸为起始原料,与2-氰基-4'-溴甲基联苯缩合制得2;2在二氯化锡的作用下变成中间体3,3与原碳酸四乙酯反应得到4,4与三丁基叠氮锡反应制得5,5水解变成6,6跟三苯基氯甲烷反应生成7,7与8反应得到坎地沙坦酯产品。其中8通过用氯甲酸乙酯和二氯亚砜在过氧苯甲酸的催化下制得9,再经与环己醇酯化、碘置换氯得到。
2 基因毒性杂质的判断
2.1基因毒性杂质的判断依据
判断一个杂质是否有基因毒性的方法有很多,为每个杂质建立起具体的实验室模型,然后进行基因毒性试验显然不现实。目前通用的判断方法之一是依据警示结构来进行。警示结构是依据化学结构的活性以及同类型化合物的性质等因素设立的,因此具有一定的科学性。虽然说相关文献给出的警示结构覆盖的范围很广,由此带来的工作量也很大,但是依据警示结构来进行判断和分析仍然是目前位置比较直观比较容易进行的一个方法。已有文献报道的警示结构有三十多个[2],每一个结构后面均代表着一系列的化合物。其中比较典型的是脂肪族卤代结构和酰卤结构,均属警示结构,具体结构如下:
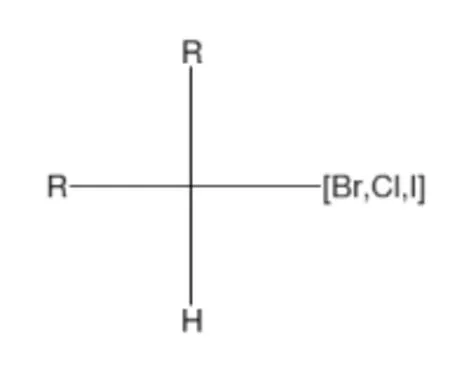
脂肪族卤代结构,R可以是任意原子或基团。
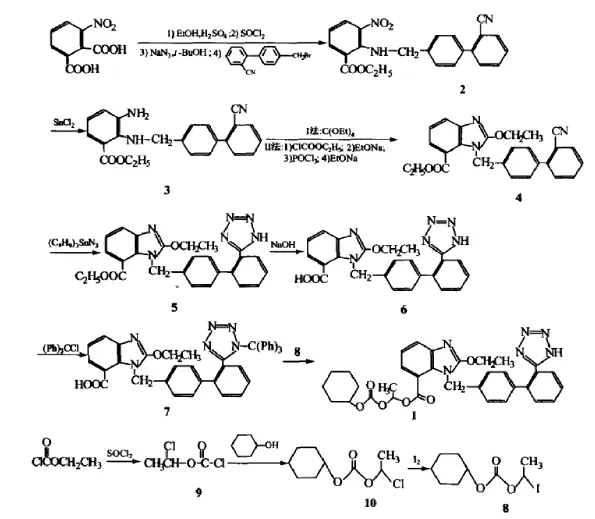
坎地沙坦酯的合成路线
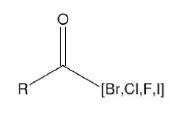
酰卤结构,R可以是除了-OH,和-SH之外的任意原子或基团。
其实这样的警示结构也很容易被人理解,这类化学结构性质活泼,如果化合物里有这样的化学结构存在,则化合物存在跟DNA发生作用可能,进而导致DNA发生变异。
2.2坎地沙坦酯基因毒性杂质分析
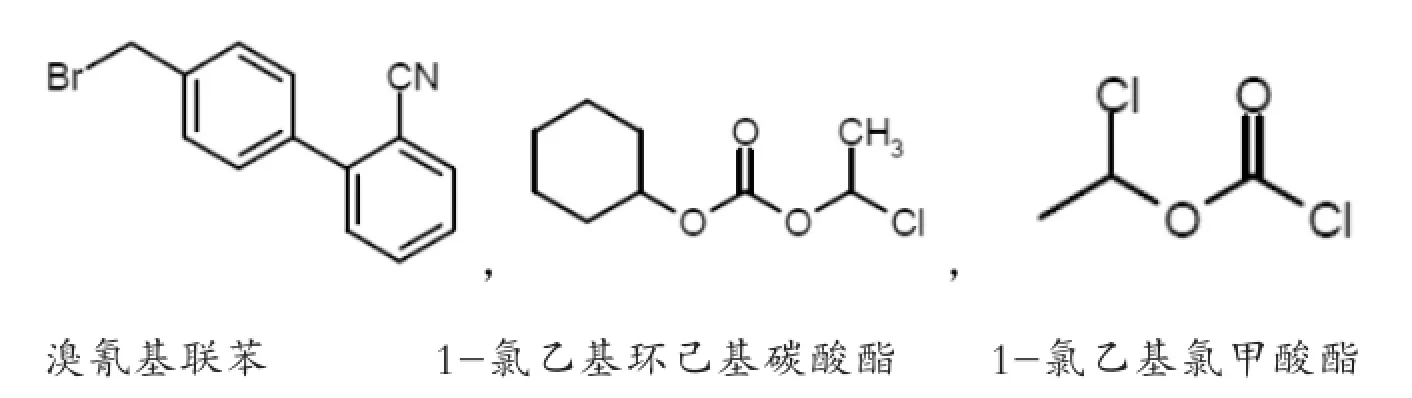
通过在坎地沙坦酯的合成工艺中查找上述两个结构,很容易就能找到其中的一些潜在基因毒性杂质,分别是:
如何对潜在基因毒性杂质进行控制呢?根据EMEA的指南[3],如果一个杂质存在“警示结构”,该杂质的致突变试验(Ames)结果为阴性时,则可以将该杂质的“基因毒性”排除,不需要进行进一步的确认研究;或者潜在基因毒性杂质水平控制在TTC(毒理学关注阈值)的水平以下时,除非是具有非常强的基因毒性的物质(N-亚硝基化合物及偶氮化合物或黄曲霉毒类化合物),可以不对该杂质进行日常的监测。上述两种方法中,进行Ames试验费时费力;而通过工艺优化等措施将这类杂质控制在TTC的水平则比较容易达成。
TTC值最初由美国FDA提出,制定TTC值是为了对任何没有得到充分研究的化学物质通常应用的接触水平作出限定,从而最大程度的阻止它们产生较大的致癌风险或其它毒性作用。就大多数药品来说,当人们每天摄入的基因毒性物质的TTC值达到1.5μg时,即被认为将存在可能致癌的风险;如果每日摄入量低于这个TTC值,那么即使终身服用,它的致癌的风险将不会高于1×10-5的概率,这一风险是可以被接受的。
通过马丁代尔大药典上的数据可以得知坎地沙坦酯的最大日服剂量为32 mg,那么根据1.5μg的基因毒性杂质摄入限度计算,坎地沙坦酯中潜在基因毒性杂质的残留不得超过46.875 ppm。溴氰基联苯为沙坦类药物合成中普遍需要的一个化合物,因此本文选取该化合物进行杂质残留的举例分析。
溴氰基联苯是起始物料之一,所以它的残留不可避免,但是在与另一起始物料反应后,即使存在少量的残留,由于其能在多种有机溶剂中溶解,在后续诸多的工序中也能除去,比如分层,离心分离,洗涤等。因此从理论上讲,它不太可能残留到最终的产品中去,而通过实际的HPLC检测,也证明了这一推论。
具体的检测方法如下:
供试品溶液:精密称取本品约50 mg,置于50mL的容量瓶中,加混合溶剂(水:乙腈=40:60)使其溶解并稀释至刻度。
以冰乙酸-水-乙腈(1∶43∶57)为流动相A;
以冰乙酸-水-乙腈(1∶10∶90)为流动相B;
色谱条件:C18柱,3.9 mm×150 mm×4μm,检测波长254 nm;流速0.8mL/min,进样体积10μL;按下表进行梯度洗脱。
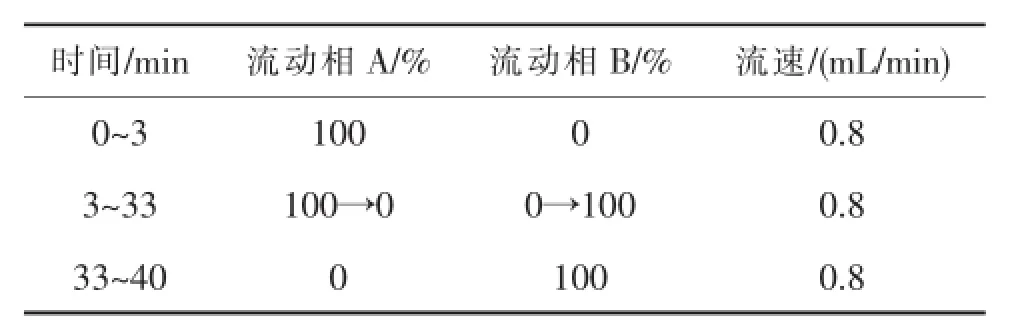
表1 梯度洗脱程序
实验结果:
溴氰基联苯的检测限为10ppm,图谱如图1:
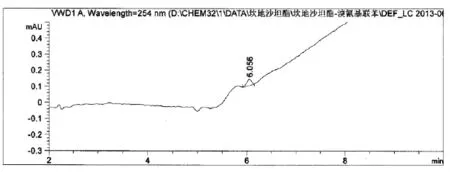
图1 氰基联苯检测限HPLC图谱
坎地沙坦酯中该杂质的检测结果:
从坎地沙坦酯的检测结果图谱中,我们可以发现该杂质未被检出。溴氰基联苯的检测限为10 ppm,所以可以定性的确定该杂质的含量不超过10 ppm,而通过TTC计算得到的控制限度为46.875 ppm,因此可以确定该杂质的残留水平符合要求,可以不进行其他的基因毒性的分析研究了。
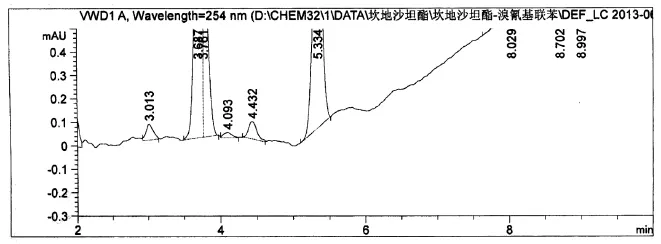
图2 坎地沙坦酯的HPLC图谱
3 结论
综上所述,通过对工艺进行分析确定出潜在基因毒性杂质,然后根据最大日服剂量和TTC计算出杂质的允许残留限度,接着通过理论和实验数据去证明该杂质的残留是符合要求的,这样的流程,虽然比较简单也不充分,但是在缺乏相关杂质的基因毒性数据的时候,其相对来说是一个比较科学的流程,也是被FDA和EMA等机构接受的一个流程。不过上述内容只是一个潜在基因毒性杂质控制的一个例子,潜在基因毒性杂质的控制,还是一个新鲜事物,如何更科学的去认识它,控制它,方法肯定不止一个。虽然国外的很多指南和方法可以拿来用,但是针对国内销售的药品,该方面的研究还鲜有报道,因此要想让国内的老百姓用上好药,用上放心药,其实还任重而道远。
[1]曹日晖,钟庆华,彭文烈,等.坎地沙坦酯的合成[J].中国医药工业杂志,2003,34(9):425-427.
[2]Romualdo Benigni,Cecilia Bossa,Nina Jeliazkova,et al. The Benigni/Bossa rulebase formutagenicity and carcinogenicity-a module of Toxtree[M].EUR 23241 EN.Italy. European Comm ission Joint Research Centre.2008:30-70.
[3]EMA/CHMP/SWP/431994/2007 Rev.3,Questions and answers on the'Guideline on the limits of genotoxic impurities'[M].London.European Medicines Agency.2010:2-6.
A Brief Analysis on Potential Genotoxic Im purities Control in Candesartan Cilexetil
QIAN Zhi-ying,ZHAO Bin-feng
(Zhejiang Kinglyuan Pharmaceutical Co.,Ltd.,Shaoxin,Zhejiang 312369,China)
The decision of potential genotoxic impurities is based on the structural alerts.After the analysis of potential geotaxis impurities arise in the synthesis route of API candesartan cilexetil,the control limits for such impuritieswere established.And then the controlling of bromine cyanobiphenyl impurity was given as an example to explain the way of potential geotaxic impurities control.
Candesartan cilexetil;genotoxic;impurity control
1006-4184(2015)6-0027-04
2015-03-03
钱志英(1977-),女,浙江绍兴人,工程师,学士学位,主要从事原料药的合成与生产。E-mail:sxqzy918@163.com。
