基于子模性质的基因表达谱特征基因提取
2015-09-11蒋智谋姚唐龙
蒋智谋++姚唐龙


摘要:针对高维小样本的特点的基因表达谱数据,提出一种基于子模性质的特征基因提取算法。首先根据图论知识将独立的基因属性转换为具有结构信息的邻接图,之后对表征基因关系的邻接矩阵利用子模性质的目标函数进行分析,事先设置特征基因子集的个数K,使用贪心算法通过迭代K个步骤,将每一次选取的特征基因加入到集合S中,作为最终选择的特征基因子集;最后,使用SVM分类器进行分类实验。通过几组公开的基因表达谱数据集的实验结果分析说明了该方法的有效性。
关键词:基因表达谱;子模;邻接矩阵;贪心算法
中图分类号:TP18 文献标识码:A 文章编号:1009-3044(2015)17-0194-03
Gene Expression Profiles Feature Extraction Based on Submodular
JIANG Zhi-mou1,2, YAO Tang-long2
(1.Anhui Lang Electronic Technology Co., Ltd, Hefei 230039, China;2.School of Electronics and Information Engineering, Anhui University, Hefei 230039, China)
Abstract: The characteristics of high-dimensional gene expression data for small samples, this paper presents a characteristic gene-based Submodular the nature of the extraction algorithm. First, according to the knowledge of graph theory to separate genes properties into adjacent graph with structural information, the following relationship for the characterization of gene adjacency matrix using the sub-mode to analyze the nature of the objective function, through pre-set number of feature gene subset, using greedy algorithm iteration steps, each one gene is added to the selected feature set, as the final selection of a subset of genes characteristic; Finally, using the SVM classifier to classify experiments. Through several sets of experimental results published analysis of gene expression data sets illustrate the effectiveness of this method.
Key words: gene expression profiling; submodular; adjacency matrix; greedy algorithm
随着新分子生物学技术和DNA微阵列技术的迅速发展,可以同时定量测量生物样本中成千上万的基因表达水平,这一技术产生的基因表达谱数据能够揭开隐含的、以前未知的生物学知识。近几年来,研究学者利用统计学和模式识别等知识对基因表达谱数据进行分析,对致病的肿瘤基因进行有效的挖掘,从而对肿瘤的类型作出准确的诊断和分类预测。
目前对高维小样本的基因表达谱数据,特征基因的子集选择有效解决了高维数据所面临的“维数灾难”问题。自1999年Golub等[1]人第一次提出了以“信噪比”作为评价指标,采用加权投票法过滤冗余基因构建分类模型之后,研究学者提出了许多新的特征基因挖掘方法。Mishra等[2]人提出一种改进的信噪比方法,Peng等[3]人提出使用互信息来度量特征之间的相关性程度选择信息基因,Mukkamala等[4]采用[t]统计量的方法过滤冗余基因,张靖等[5]提出一种改进的Lasso方法迭代剔除冗余的基因,Xu等[6]提出一种基于标准差分布差异(SDED)选择特征基因的方法,Hang等[7]人提出一种基于稀疏表示的肿瘤基因表达谱数据分类方法。
传统特征基因选择方法大部分是对基因进行独立分析,没有考虑基因之间的相关信息,因此,该文提出一种基于子模性质的特征基因提取算法。首先将全部基因看作高维空间中的一组点集构建邻接图,然后利用具有子模性质的目标函数迭代的选取特征基因加入到基因子集中,得到最终的特征基因子集,最后利用SVM分类器进行分类。通过对公开的白血病和前列腺癌数据集的实验结果分析,验证了该文方法的可行性和有效性。
1 数据处理与算法分析
通常使用[G=g1,g2,...,gN]表示一个样本中所有的基因构成的集合,每一行表示一个样本,每一列表示一个基因。其中[gi(1≤j≤N)]代表第[j]个基因,[N]表示基因的总个数。[S=si,s2,...,sM]表示[M]个样本集合,[M]为样本的个数,一般有[M≤N]。
[G=g1,1g1,2…g1,Ng2,1g,2…g2,N????gM,1gM,2…gM,N] (1)
其中:[gi,j]表示基因[gj]在样本[si]中的表达值。
1.1 子模的性质与选择
考虑有限集合[V]和定义在其幂集[2E]的一个实函数[f:2V→R],当且仅当对于任意的集合[S?V],[T?V]满足:
[f(S?T)+f(S?T)≤f(S)+f(T)] (2)
则称函数[f(?)]为具有子模性质的目标函数,称之为子模函数(Submodular function)。子模具有凸面离散模拟的性质,这种性质使得一个连续函数能够得到局部最优解,近几年来子模在组合优化问题中求解[f]最大值问题起了至关重要的作用,且子模具有增益递减的性质,即对任意的[R?S?V]和[s?V],有:
[f(S?s)-f(S)≤f(R?s)-f(R)] (3)
子模特征选取的最终目标是在整个集合[V]中选择一个子集[S]使目标函数达到最大化,且使得子集[S]的大小不超过[K] ([K]为需要选取的特征子集的个数,在初始化时设置),这种约束方式对求[f]的最大值问题能够达到较好的结果。该文使用算法一个给定约束为:
[S≤K] (4)
其中[S]为集合[S]的势,即从所有特征集合[V]中选取的特征子集[S]的个数,[K]为初始时设定的目标值。综上所述,子模的最大化问题可以描述为公式(5)所示:
[maxS?Vf(S):S≤K] (5)
1.2 基于图的子模特征提取
设[G=g1,g2,???,gN]表示一个样本中所有基因构成的集合,其中[gj(1≤j≤N)]表示第[j]个基因,[N]表示基因个数。根据图论知识将基因表达谱数据中每一个基因映射为高维空中的一个节点,以基因点集通过构建权重图[G(V,E)],其中[V=1,2,...N]为基因点集,[E]表示节点之间的边集,使用非负的权值矩阵[wi,j] 来连接每一条边[(i,j)]并赋权值,目标是在整个基因集[V]中找到一个最优的子集[S]。为了利用子模的性质,引入了式(6)作为子模目标函数:
[f(S)=i∈Vmaxj∈Swi,j] (6)
该目标函数为归一化的子模目标函数,是经典的无容量限制的设施选址(Uncapacitated Facility Location Problem)位置函数[8]。在式(6)中,由于[wi,j≥0] 恒成立,因此该目标函数满足增益递减的性质,可以利用贪心算法[9]获得该目标函数近似最优解。对目标函数使用贪心算法求解特征子集的过程如图1所示:
[贪心算法求解[f]的过程如下:\&输入:[G(V,E)]的边[(i,j)]的权重值矩阵[wi,j]
[K]:选取的特征基因个数,即[S]
输出:选取的特征基因子集[S]
1) 初始化[S=?],[ρi=0,i=1,2,...,N],其中[N=V];
2) 当[S>K]时,执行步骤3,否则执行步骤4;
3) [k*=argmaxk∈V\Si∈V,(i,k)∈E(maxρi,wi,k-ρi)]
[S=S?k*]
对所有的[i∈V]有:[ρi=max(ρi,wi,k*)];
4) 当[S>K]时,结束。\&]
图1 子模函数提取特征子集算法
首先固定特征基因子集的大小[K],[wi,j]是由基因表达谱经过预处理之后的基因属性构成的邻接矩阵,初始时[S=?],然后根据子模态目标函数的性质,利用贪心算法通过迭代[K]步,每经过一次计算之后将一个新的元素加入到子集[S]中,最终得到特征子集[S],且[S=K]。
1.3 算法步骤
该文通过将基因看作高维空间中的节点进行构图,然后利用子模性质的目标函数选取特征基因,算法的主要步骤如下:
1) 对基因表达谱数据[G]进行无关基因的过滤,过滤之后的基因表达谱数据为[G']。
2) 对基因表达谱数据[G']按照(7)式构造图的邻接矩阵,并进行高斯权赋值:
[W=(wi,j)=exp(-di-dj22σ2) ,i≠j0 ,i=j] (7)
该文中的实验统一设置[σ]的值为[σ=mean(G'(:))],即所有样本基因的均值。
3) 根据图1的算法从基因集合[V]中选取特征基因子集[S],根据分类精度最终确定最优的特征子集合的数目。
4) 使用10折的交叉验证(10-fold cross-validation),输入线性核SVM分类器进行分类,实验运行5次,最终取5次的平均值作为分类精度。
2 实验结果与分析
作者对急性白血病和前列腺癌两类公开的基因表达谱数据集进行了实验,其中白血病数据集含有52个样本(24个为急性淋巴性白血病—ALL,28个为急性粒性白血病—AML),每个样本有11225个基因;结肠癌数据集中含有102个样本,其中有52例样本为前列腺肿瘤(Prostate tumor)样本,50例样本为正常组织(normal tissues)样本。每个样本中包含10509个基因。
根据1.3节描述的算法步骤,对白血病和前列腺癌数据集进行实验验证,实验结果如表1所示:
根据表1的结果可知实验中白血病数据的平均分类准确率为100%,而选取的特征基因数只有7个特征基因,结肠癌数据的最高分类准确率为96.08%,最终的平均分类准确率达到94.90%,平均只选取19.6个基因,都取得了较好的实验结果。在每一次实验中,选取特征基因的数量差别不大,说明基于子模选取的特征基因子集具有一定的稳定性和较好的区分肿瘤类型的能力。
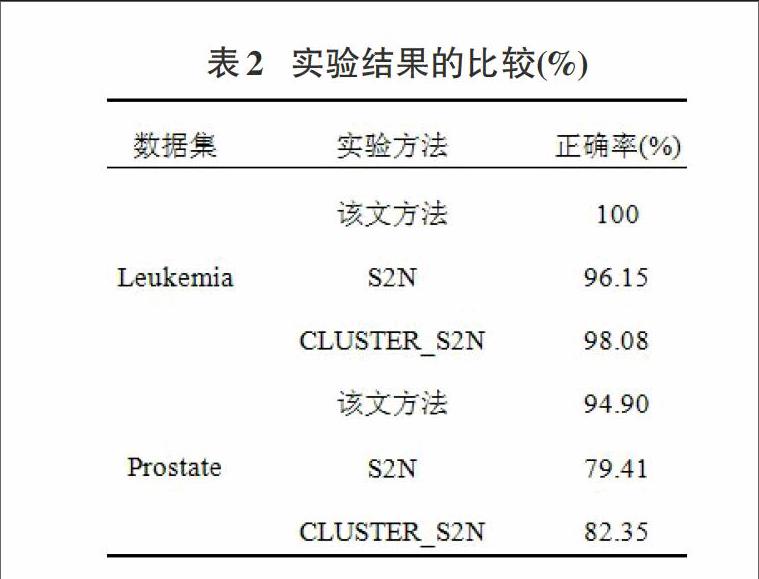
按照同样的实验步骤,将该文的特征基因提取方法与以“信噪比”为指标的特征基因提取方法和文献[10]中提出的CLUSTER_S2N特征基因提取方法进行实验比较,采用线性核SVM分类器,使用平均正确率作为最终的实验结果,实验结果如表2所示:
由表2可知,在相同实验条件下,根据最终的识别正确率来说,该文提出的方法相较于其他两种特征基因提取方法正确率高,具有一定优势,表明基于子模的特征提取方法能够过滤掉噪声和无关基因,选取的特征基因子集为与肿瘤类型相关的基因,因此能够得到较好的识别率。这是由于该文方法利用了基因之间的相关信息,CLUSTER_S2N方法虽然使用了聚类的思想,但选取的基因子集冗余度较高,而S2N方法每次只对单个的基因进行处理,忽略了基因之间的相关性,因此对前列腺癌数据识别正确率不高。
3 结束语
为了对基因表达谱数据的特征基因有效挖掘,许多特征基因提取方法相继被提出。作者提出一种基于子模性质的特征提取方法应用在基因表达谱的特征基因提取中,实验结果表明基于子模性质提取的特征基因子集达到了较高的正确率,优于传统的特征提取方法,在以后的研究中可以通过优化子模目标函数得到更高的识别率。
参考文献:
[1] Golub T R, Slonim D K, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring[J]. science, 1999, 286(5439): 531-537.
[2] Mishra D, Sahu B. Feature selection for cancer classification: A signal-to-noise ratio approach[J]. International Journal of Scientific & Engineering Research, 2011, 2(4): 1-7.
[3] Peng H, Long F, Ding C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy[J]. Pattern Analysis and Machine Intelligence, IEEE Transactions on, 2005, 27(8): 1226-1238.
[4] Mukkamala S, Liu Q, Veeraghattam R, et al. Feature selection and ranking of key genes for tumor classification: using microarray gene expression data[M]//Artificial Intelligence and Soft Computing–ICAISC 2006. Springer Berlin Heidelberg, 2006: 951-961.
[5] 张靖,胡学钢,李培培,等. 基于迭代Lasso的肿瘤分类信息基因选择方法研究[J]. 模式识别与人工智能,2014,(1).
[6] Xu W, Wang M, Zhang X, et al. SDED: A novel filter method for cancer-related gene selection[J]. Bioinformation, 2008, 2(7): 301.
[7] Hang X, Wu F X. Sparse representation for classification of tumors using gene expression data[J]. BioMed Research International, 2009.
[8] Korupolu M R, Plaxton C G, Rajaraman R. Analysis of a local search heuristic for facility location problems[J]. Journal of algorithms, 2000, 37(1): 146-188.
[9] Jain K, Mahdian M, Saberi A. A new greedy approach for facility location problems[C]//Proceedings of the thiry-fourth annual ACM symposium on Theory of computing. ACM, 2002: 731-740.
[10] 阮晓钢, 晁浩. 肿瘤识别过程中特征基因的选取[J]. 控制工程, 2007, 14(4): 373-375.
