基于结构和药效团特征的人类腺苷受体拮抗剂选择性比较
2015-09-03曾凌晓李欣然金宏威刘振明张亮仁
曾凌晓 李欣然 金宏威 刘振明 张亮仁
(北京大学药学院,天然药物及仿生药物国家重点实验室,北京 100191)
基于结构和药效团特征的人类腺苷受体拮抗剂选择性比较
曾凌晓 李欣然 金宏威 刘振明*张亮仁*
(北京大学药学院,天然药物及仿生药物国家重点实验室,北京 100191)
腺苷受体是重要的治疗靶标,选择性腺苷受体拮抗剂具有广泛的临床应用前景.本文通过同源模建构建了腺苷A1、A2B和A3受体的结构,采用LigandScout 3.12软件分别构建了腺苷受体四种亚型的拮抗剂药效团模型.然后利用Schrödinger程序中的Induced Fit Docking模块完成受体-拮抗剂结合模式的预测,并与药效团结果进行比对.结果发现,由于结合口袋部位的残基在家族间高度保守,模建得到的各个亚型受体的初始结构活性口袋部位极为相似,无法用于亚型选择性拮抗剂的识别.而腺苷受体四种亚型拮抗剂药效团的药效特征与空间排布都不同,并与以前突变实验信息相吻合.研究结果说明,结合口袋部位的优化是模建中的关键步骤,基于配体的药效团模型所包含的一系列药效特征元素如氢键受体、氢键供体、疏水基团、芳环中心,可以很好地表征受体结合部位氢键、疏水空腔的位置及其方向.本文研究结果可以为进一步的优化同源模建结果,寻找新型的人类腺苷受体选择性拮抗剂提供理论依据.
药效团; 人类腺苷受体; 拮抗剂; 选择性
1 引 言
G蛋白偶联受体(GPCRs)是一个庞大的跨膜蛋白受体家族,目前已经发现800多个成员,主要分为5个亚家族:谷氨酸、视紫红质、粘附、Frizzled/ Taste2以及分泌素.1它们的共同结构特征是肽链由N末端、7个跨膜α螺旋(TM1–TM7)、C末端、3个胞外环(ECL1–ECL3)及3–4个胞内环(ICL1–ICL4)组成.GPCRs可以识别多种胞外配体,激活信号转导通路,引发细胞内反应.2GPCRs参与的信号传导过程广泛调控感知、生殖、发育、生长、代谢等多种生理过程,与糖尿病、心脏病、肿瘤、免疫和感染性疾病、精神疾病等重要疾病的发生、发展及治疗密切相关;3鉴于GPCRs在生理病理过程中的重要作用,其作为重要的“药靶”一直以来在新药研发领域受到广泛关注,市场上30%–40%临床用药物都作用于GPCRs.4
腺苷是一类重要的内源性核苷,参与包括核酸合成、氨基酸代谢、细胞代谢调节在内的多个关键生命过程,其生物学调控异常会导致心血管系统、中枢神经系统、呼吸系统和胃肠道系统等多种疾病.5腺苷在生物体内的诸多生理作用是由腺苷介导的,其作为信号分子激活腺苷受体,产生相应生理作用.腺苷受体属于GPCRs超家族,在人体组织中分布广泛.6目前已经发现了腺苷受体的4种亚型,并已被成功克隆,分别为:A1、A2A、A2B和A3.7,8腺苷受体是重要的治疗靶标,选择性腺苷受体拮抗剂具有众多治疗前景,包括心血管疾病、炎症和神经退行性疾病.9然而,在临床应用中针对腺苷受体药物研发进展缓慢,目前已知的腺苷受体拮抗剂发挥抑制作用的过程中,大部分都存在选择性差的缺陷,由此导致临床应用中不良反应的发生.5因此,针对腺苷受体选择性拮抗剂的研究就显得尤为重要,研究腺苷受体拮抗剂的特异性和选择性对进一步阐明其作用机理和开发新拮抗剂均有重要意义.
计算机辅助药物设计方法从方法学上主要分为两种:基于受体的药物设计和基于配体的药物设计.10晶体结构是目前研究者了解GPCRs功能和进行调控分子设计的基础,第一个被解析的GPCRs晶体结构是牛视紫红质.11由于结构生物学技术的快速发展,科学家开发出新的方法来解决膜蛋白表达、溶解和结晶问题的瓶颈,越来越多的GPCRs晶体结构得到解析.1但从整个GPCRs家族来看,目前解析出来的晶体结构数量还是太少,对于腺苷受体家族,目前也只有腺苷A2A受体有晶体结构解析出来.因此大部分GPCRs靶蛋白的研究都是基于同源建模获得的结构而进行的.12属于同一亚家族的不同亚型有着不同的氨基酸序列、组织分布和药理作用,但与此同时结合口袋部位的残基往往在家族间高度保守.因此,模建具有受体亚型选择性的结构从而进行选择性配体的设计仍然是一个挑战.13
相比于晶体结构数据的缺乏,GPCRs具有丰富的配体数据信息.药物分子在体内发挥生理作用是与生物大分子相互作用的结果,受体与配体通过特定的结合部位产生相互作用.因此,特定的三维空间构象对于分子呈现其药效活性具有很重要的意义.从生物活性小分子的三维构象出发分析药物与受体的相互作用模式,是药物分子设计与选择性分析常用的策略.14基于配体的药效团设计是在靶标蛋白结构信息缺少时进行药物设计的主要手段,其基本流程为:根据化合物结构特征的空间排列形式进行分子叠合,得到对同一靶标位点进行识别,表现出相似药效特性的一类分子所具有的共同特点.15而药效团所包含的药效特征元素如氢键受体(HBA)、氢键供体(HBD)、疏水基团(HY)、芳环中心(RA),能够反映出受体结合部位对应的特征元素.
腺苷受体是一类研究相对成熟的GPCRs蛋白,包括本课题组在内,很多研究小组都对它们的调控分子进行了广泛的研究,并发现了多个系列的腺苷受体亚型拮抗剂.研究发现腺苷受体不同亚型调控分子的结构之间存在着一些明显的特征差异,考虑到小分子与受体结合时的互补和契合,这种差异是否也反映了结合状态下受体蛋白空间拓扑结构的特征呢? 本文拟通过基于配体结构的药效团和同源模建腺苷受体四种亚型的结构,比较分析腺苷受体不同亚型之间选择性拮抗作用的内在规律.
2 计算方法
2.1 同源模建
腺苷A1、A2B和A3受体同源模建所用的模板是A2A受体的晶体结构(PDB ID:4EIY),来源于PDB蛋白晶体结构数据库(www.rcsb.org).16A1,A2B和A3受体的序列(A1ID:P30542,A2BID:P29275,A3ID:P33765),均来自SWISS-PROT数据库.17同源模建均采用Accelrys公司的Discovery Studio 2.5(DS 2.5)18中的MODELER模块进行搭建,计算中选用的各项参数除特别说明外均使用缺省值.
2.1.1 序列比对
首先对PDB蛋白晶体结构数据库中得到的腺苷A2A受体晶体结构做了初步处理,具体包括:删除T4溶菌素蛋白,删除水分子,删除原始小分子配体,添加氢原子.序列比对采用的是DS 2.5中Align Sequence to Templates模块,将A1,A2B和A3受体的氨基酸序列与处理后的模板蛋白A2A受体进行比对,确定模板蛋白与目标蛋白氨基酸序列之间的残基匹配情况.
2.1.2 模型的构建
根据序列比对的结果,以腺苷A2A受体的蛋白拮抗状态晶体结构为模板,采用DS 2.5中的Build Homology Models模块分别对A1,A2B和A3进行三维结构同源模建,由程序自动生成10个模型,选取概率密度函数(PDF)对蛋白质几何性质打分最高的模型.
2.1.3 模型的评价
得到的模型采用PROCHECK程序19进行合理性评价,PROCHECK主要用于评价模型中残基与残基之间的立体化学性质,考察残基之间的φ和ψ两个角度分布在Ramachandran图中的分布是否合理.Ramachandran图显示了各个残基间的φ和ψ这两个角度是否出现在合理的区域.20在Ramachandran图中,共有四个区:最佳合理区(the most favored regions),额外合理区(additional allowed regions),一般合理区(generously allowed regions),不合理区(disallowed regions),以模板中的氨基酸在这四个区域分布的百分比来评价模型的好坏.
2.2 训练集的选择
从ChEMBL数据库中(https://www.ebi.ac.uk/ chembl)21收集腺苷受体四个亚型的拮抗剂.使用本实验室夏杰博士建立的protocol流程在Pipeline Pilot 7.5软件中对所有配体进行拓扑相似性比较,22,23以结构多样性为选择原则,针对每一个腺苷受体亚型分别保留6个拮抗剂用于下一步的药效团模建.24
2.3 药效团模型的构建
使用LigandScout 3.12软件,25采用Omega-best方法,针对每个配体分子得到一组在合理的能量范围内具有一定代表性的化合物构象.采用默认参数建立了基于配体的药效团模型,根据Pharmacophore-Fit打分函数,最终挑选每个亚型打分最高的模型作为最终的药效团模型.
2.4 分子对接
针对腺苷受体四种亚型,挑选出与最优药效团模型匹配最好的配体分子,与对应蛋白结构进行分子对接.
分子对接的过程采用Schrödinger程序,26首先采用Protein Preparation Wizzard模块对蛋白进行预处理(包括核准键级、查找残基及原子重叠、生成氢键及能量最小化等工作);随后使用LigPrep模块分别对分子进行能量最小化(使用OPLS_2005力场)预处理;最后使用Induced Fit Docking模块进行诱导对接.
3 结果与讨论
3.1 同源模型的评价
选择了腺苷A2A受体的拮抗状态晶体结构(PDB ID:4EIY)作为同源模建的模板蛋白,序列比对结果如图1所示.腺苷A1受体与腺苷A2A受体的相似度(similarity)为69.2%,一致性(identity)为51.3%;腺苷A2B受体与腺苷A2A受体的相似度为77.2%,一致性为60.6%;腺苷A3受体与腺苷A2A受体的相似度为66.1%,一致性为41.3%.
使用PROCHECK程序评价模型结构的立体化学参数,此程序根据经验比较所给蛋白质结构与最合理的蛋白质结构之间立体化学性质的差异.通过程序生成的Ramachandran图来表示所有氨基酸残基骨架的二面角分布(表1,图2).结果显示,三个模建受体均没有残基处于不合理区域,因此通过同源模建获得的腺苷A1、A2B和A3受体模型从总体上看结构是合理的.
对于GPCRs的模建,由于都具有七次跨膜的保守性结构,主链的模建并不困难.由上述结果也能反映出模建初始模型在结构合理性上达到要求.同源模建主要应用于分子对接和虚拟筛选,为得到选择性腺苷受体拮抗剂,要求模型具有结构上的选择性区分,这种差异主要体现在活性口袋关键氨基酸残基的位置和角度上.然而当我们比对模建的腺苷受体与初始模板的活性口袋残基时,发现模建得到的腺苷受体三个亚型结构与初始模板蛋白非常相似,腺苷A1、A2B和A3受体模型与初始模板活性口袋残基叠合均方根偏差(RSMD)分别为0.0087、0.0098和0.0072 nm(图3).

图1 腺苷A1(A),A2B(B)和A3(C)受体与腺苷A2A受体序列比对Fig.1 Sequence alignment of A2Aand A1(A),A2B(B),A3(C) adenosine receptors(AR),respectively

表1 腺苷受体模型PROCHECK评价结果Table 1 Results of PROCHECK of modeled adenosine receptor
这种活性口袋相似性的主要原因是,属于同一亚家族的不同亚型的蛋白,其结合口袋部位的残基往往在家族间高度保守.13从图4可以看出,腺苷受体家族4个亚型的配体结合口袋残基保守性较高,核心的配体–受体相互作用包括配体芳环体系与Phe1685.29(上标代表氨基酸残基在每个螺旋区和环区相对最保守的位置,Ballesteros-Weinstein编号法27)侧链的π–π堆叠作用,Leu2496.51,Ile2747.39,Met1775.38的疏水相互作用,以及Asn2536.55的氢键作用,这些残基对配体结合作用起到主要贡献.这种残基之间的相似性为模建和基于受体–配体间相互作用设计选择性调控分子带来了困难.这也表明,不经过优化的同源模建粗模型即使满足结构合理性,但与真实情况存在差距.因此,模建具有受体亚型选择性的结构从而进行选择性配体的设计仍然是一个挑战,需要进一步从配体出发分析不同亚型拮抗剂的分子结构和药效团特征,进而获得可信的具有选择性的模型结构.
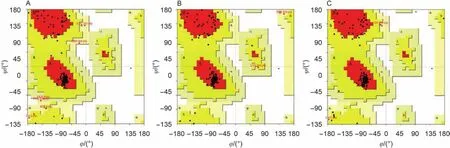
图2 腺苷受体模型的Ramachandran图Fig.2 Ramachandran plots of modeled adenosine receptors

图3 腺苷A2A受体活性口袋(A)及模建的腺苷A1受体(B)、腺苷A2B受体(C)、腺苷A3受体(D)与初始模板的结合口袋残基叠合图Fig.3 Binding pocket residues of A2AAR(A) and superimposition of binding pocket residues of the homology models of A1AR(B),A2BAR(C),A3AR(D) with initial template structure

图4 腺苷受体四种亚型配体结合口袋残基比较Fig.4 Residue variations in the ligand-binding pocket between four adenosine receptor subtypes Residues are colored according to their conservation:red,fully identical in all 4 subtypes;green,in 3 subtypes;blue,in 2 subtypes;purple,in only one subtype
3.2 训练集的选择
从ChEMBL数据库所挑选的高选择性拮抗剂中,根据配体多样性筛选原则针对腺苷受体四种亚型分别构建含有6个化合物的训练集(见图5–图8),训练集化合物活性值(Ki)见表2–表5.28–48

图5 应用于训练集的高选择性腺苷A1受体拮抗剂的化学结构Fig.5 Chemical structure of highly selective A1AR antagonists for training set

表2 腺苷A1受体训练集化合物实测生物活性值列表Table 2 Experimental biological activity data of training set of A1AR
药效团模型中两个重要药效特征(氢键供体和氢键受体)表征配体和受体相互识别的氢键相互作用,疏水特征可表征药物非极性区域与受体的非极性区域之间的疏水相互作用,芳香环主要参与药物分子与受体中π电子离域系统相互作用,药效团中芳香环特征表征这一作用.

图6 应用于训练集的高选择性腺苷A2A受体拮抗剂的化学结构Fig.6 Chemical structure of highly selective A2AAR antagonists for training set

表3 腺苷A2A受体训练集化合物实测活性值列表Table 3 Experimental biological activity data of training set of A2AAR

图7 应用于训练集的高选择性腺苷A2B受体拮抗剂的化学结构Fig.7 Chemical structure of highly selective A2BAR antagonists for training set
3.3 药效团模型比较
采用LigandScout 3.12软件中基于配体的药效团模块构建每个腺苷受体亚型的药效团模型,结果显示腺苷受体四种亚型的拮抗剂均以氢键受体(HBA)、氢键供体(HBD)、疏水基团(HY)、芳环中心(RA)为药效团的基本药效元素.其中,RA是腺苷受体四个亚型药效团共有的药效特征,并且都与各亚型高选择化合物的杂环母核相对应.这与图4中腺苷受体四个亚型配体结合口袋的保守氨基酸残基相一致,处于5.29位置均为Phe残基,腺苷A1受体的Phe1715.29,腺苷A2B受体的Phe1735.29,腺苷A3受体的Phe1685.29与腺苷A2A受体的Phe1685.29对应;在已解析的腺苷A2A受体晶体表明该位置的Phe残基与配体形成π–π堆叠作用.49

表4 腺苷A2B受体训练集化合物实测活性值列表Table 4 Experimental biological activity data of training set of A2AAR

图8 应用于训练集的高选择性腺苷A3受体拮抗剂的化学结构Fig.8 Chemical structure of highly selective A3AR antagonists for training set

表5 腺苷A2受体训练集化合物实测活性值列表Table 5 Experimental biological activity data of training set of A3AAR
但总体来讲,腺苷受体四个亚型药效团的药效特征与空间排布都不同.腺苷A1受体拮抗剂与受体活性位点之间的相互作用形式主要包含2个氢键受体、1个疏水中心和2个芳香环.点突变实验表明,His2516.52、Thr913.36对拮抗剂的结合有着重要作用,与这里的药效团的氢键作用相对应.50,51
腺苷A2A受体拮抗剂与受体活性位点之间的相互作用形式主要包含2个氢键受体、1个氢键供体、1个疏水中心和3个芳香环,药效特征与已知晶体结构受体–配体相互作用匹配良好.Phe1685.29和Asn2536.55在A2A拮抗剂和受体之间相互作用起着关键作用.52除了上面提到的保守氨基酸残基Phe1685.29与配体的π–π堆叠作用和芳香环特征相匹配,杂环母核上的氢键供体特征与保守氨基酸残基Asn2536.55的极性相互作用相匹配.呋喃环存在与Trp2466.48的疏水作用以及与His2506.52的π–π堆叠作用.
腺苷A2B受体拮抗剂与受体活性位点之间的相互作用形式主要包含2个氢键受体、1个疏水中心和2个芳香环.研究表明,氨基酸His2516.52、Trp2476.48与配体之间存在疏水作用,这与药效团的疏水特征相对应.His2807.43和Asn2827.45与配体之间存在极性相互作用来稳定配体,与药效团的氢键受体特征相对应.53,54与其他亚型药效特征不同的一点是,腺苷A2B受体药效团模型配体的取代基部分包含一个氢键受体的药效特征,与最佳匹配化合物的羰基相对应.这一特殊的药效特征与文献中报道的非保守氨基酸残基Asn1865.42相对应,也说明此处是腺苷A2B受体选择性配体的独有特征.54,55
腺苷A3受体拮抗剂与受体活性位点之间的相互作用形式主要包含2个氢键受体、1个氢键供体、1个疏水中心和2个芳香环.且每个药效团与该受体的高活性高选择性拮抗剂分子匹配均较吻合.氢键供体的药效特征,与文献报道残基Ser2717.42和Trp943.36与配体产生氢键作用一致.疏水作用与Phe2396.44残基相对应.56
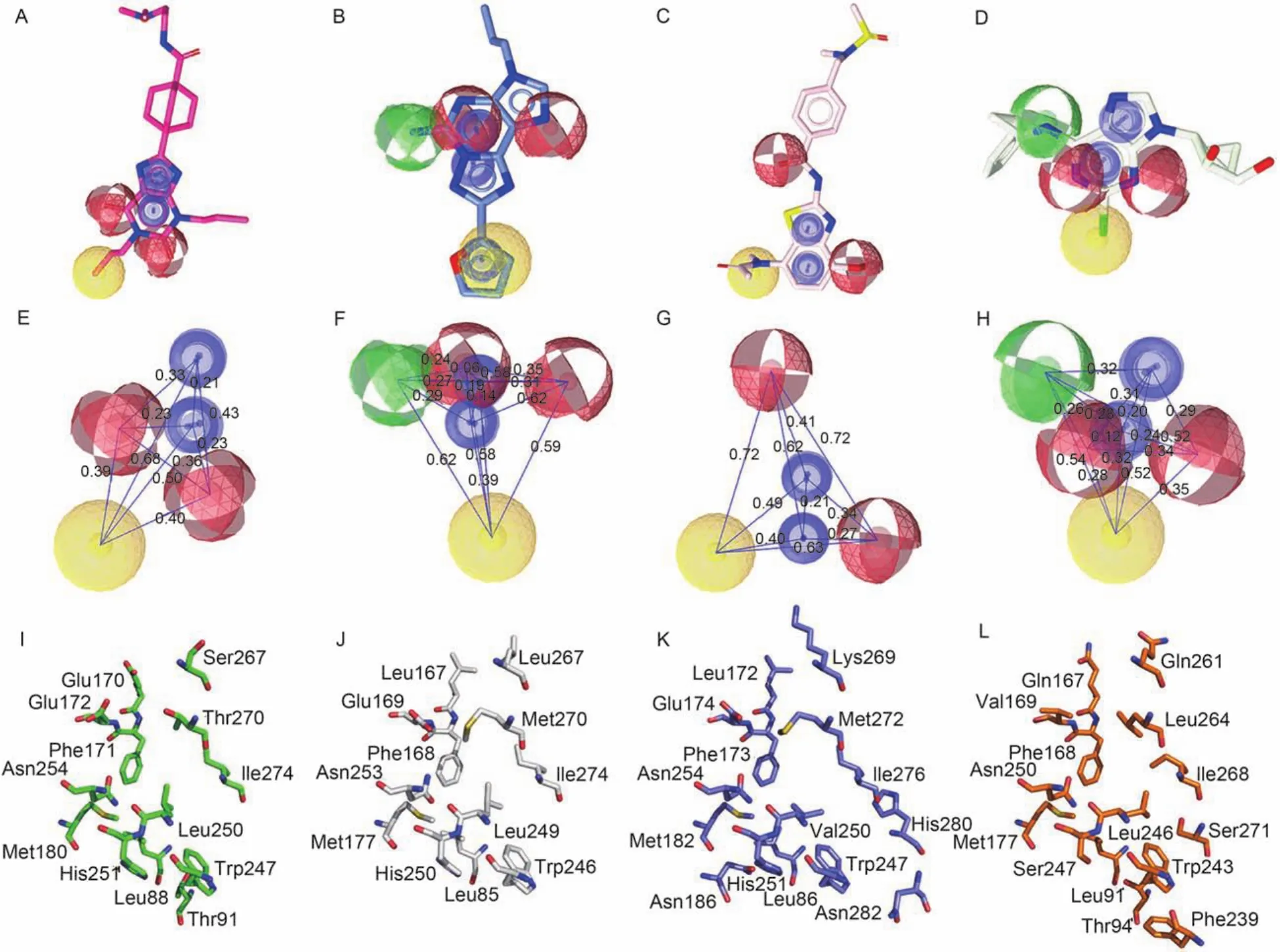
图9 腺苷受体四种亚型的最优药效团模型及对应结合口袋残基Fig.9 The best pharmacophore models and corresponding ligand binding residues of four adenosine receptor subtypes(A,E,I) A1AR;(B,F,J) A2AAR;(C,G,K) A2BAR;(D,H,L) A3AAR.distance in nm
腺苷受体四亚型拮抗剂药效团的空间距离和角度图如图9(E–H)所示,结果显示四类药效团不仅如上讨论在药效特征上存在差别,并且药效团的空间分布也不尽相同.由此可以看出,各药效基团之间均需满足一定的空间限制,从而产生选择性拮抗腺苷受体的活性作用.
与药效团对应的腺苷受体四亚型活性口袋氨基酸残基分布如图9(I–L)所示,结果发现腺苷A2A受体拮抗剂药效团模型与拮抗状态的晶体结构匹配良好,而以腺苷A2A受体为模板模建得到的其他三个亚型受体初始结构的关键氨基酸残基分布与对应药效团模型存在差异,并且与模板蛋白叠合表现出的高度相似性都表明模建后的初始模型并不能反映腺苷受体家族各亚型之间结构上选择性的差异,需要进一步对关键氨基酸残基的位置进行优化.

图10 腺苷受体四种亚型拮抗剂与对应蛋白结构相互作用图Fig.10 Interaction diagram of antagonists of four adenosine receptor subtypes binding to corresponding structures(A) A1AR;(B) A2AAR;(C) A2BAR;(D) A3AR.The antagonists are displayed in two-dimensional chemical structure and the residues of protein are shown in circle.
3.4 腺苷受体与拮抗剂作用模式分析
将腺苷受体各亚型选择性拮抗剂柔性对接到对应蛋白结构中,对接后蛋白与配体作用模式如图10所示.结果显示,各个亚型的腺苷受体与拮抗剂作用模式与上面得到药效团特征相匹配,四种亚型拮抗剂母核上的氢键作用和π–π堆叠作用与四种药效团中的氢键供体、氢键受体以及芳香环的药效特征相一致.然而,对接结果与药效团结果还是存在一些差异.对接模式中显示的氢键作用大多与保守残基如Asn、Phe相关,而将药效团结果显示出的特异性残基的氢键作用在对接结果中没有体现出来,如对接结果中腺苷A1受体与活性口袋深处残基并没有显示出氢键相互作用.这意味着同源模建粗模型直接用于分子对接可能存在偏差,有些选择性差异不能很好地体现出来.
值得注意的一点是,由于在构建药效团时挑选的是结构多样的训练集分子,这种结构的差异性使得分子的取代基部位很难进行叠合显示出共同的药效特征,而另一方面对接结果展示出了亚型特有拮抗剂的化学修饰对选择性的影响,如研究表明在黄嘌呤的8位用芳基取代提高化合物对腺苷A2B受体的选择性,57对接结果显示取代基上的苯环与非保守性氨基酸Lys2697.32有着π–π堆叠作用(图10C),这在一定程度上可以解释取代基的改变对结合选择性的影响.
4 结 论
本文基于受体结构和配体药效团特征对人类腺苷受体四种亚型拮抗剂进行比较,分析其选择性原因.结果发现,腺苷受体四种亚型拮抗剂药效团的药效特征与空间排布都不同,并与以前突变实验信息相吻合.而由于结合口袋部位的残基在家族间高度保守,模建得到的各个亚型受体的初始结构活性口袋部位极为相似,将同源模建粗模型直接用于分子对接存在偏差,有些选择性差异不能很好地体现出来.
研究结果说明,结合口袋部位的优化是模建中关键步骤,基于配体的药效团模型所包含的一系列药效特征元素如氢键受体、氢键供体、疏水基团、芳环中心,可以很好地表征受体结合部位氢键、疏水空腔的位置及其方向.本文研究结果可以为进一步的优化同源模建结果,寻找新型的人类腺苷受体选择性拮抗剂提供理论依据.
(1)Krishnan,A.;Almen,M.S.;Fredriksson,R.;Schioth,H.B.PLoS One 2012,7(1),e29817.
(2)Lappano,R.;Maggiolini,M.Nat.Rev.Drug Discov.2011,10(1),47.doi:10.1038/nrd3320
(3)Liang,F.;Yue,J.;Wang,J.;Zhang,L.;Fan,R.;Zhang,H.;Zhang,Q.Med.Oncol.2015,32(3),49.doi:10.1007/s12032-015-0486-1
(4)Overington,J.P.;Al-Lazikani,B.;Hopkins,A.L.Nat.Rev.Drug Discov.2006,5(12),993.doi:10.1038/nrd2199
(5)Chen,J.F.;Eltzschig,H.K.;Fredholm,B.B.Nat.Rev.Drug Discov.2013,12(4),265.doi:10.1038/nrd3955
(6)Ke,Y.R.;Jin,H.W.;Liu,Z.M.;Zhang,L.R.Acta Phys.-Chim.Sin.2010,26(10),2833.[柯艳蓉,金宏威,刘振明,张亮仁.物理化学学报,2010,26(10),2833.] doi:10.3866/PKU.WHXB20100916
(7)Robeva,A.S.;Woodard,R.L.;Jin,X.;Gao,Z.;Bhattacharya,S.;Taylor,H.E.;Rosin,D.L.;Linden,J.Drug Develop.Res.1996,39(3–4),243.
(8)Fredholm,B.B.;AP,I.J.;Jacobson,K.A.;Klotz,K.N.;Linden,J.Pharmacol.Rev.2001,53(4),527.
(9)Jacobson,K.A.;Gao,Z.G.Nat.Rev.Drug Discov.2006,5(3),247.doi:10.1038/nrd1983
(10)Sliwoski,G.;Kothiwale,S.;Meiler,J.;Lowe,E.W.,Jr.Pharmacol.Rev.2014,66(1),334.
(11)Congreve,M.;Langmead,C.J.;Mason,J.S.;Marshall,F.H.J.Med.Chem.2011,54(13),4283.doi:10.1021/jm200371q
(12)Costanzi,S.Methods Mol.Biol.2012,857,259.
(13)Katritch,V.;Kufareva,I.;Abagyan,R.Neuropharmacology 2011,60(1),108.doi:10.1016/j.neuropharm.2010.07.009
(14)Güner,O.F.Pharmacophore Perception,Development,and Use in Drug Design;International University Line:San Diego,2000;Vol.2.
(15)Khedkar,S.A.;Malde,A.K.;Coutinho,E.C.;Srivastava,S.Med.Chem.2007,3(2),187.doi:10.2174/157340607780059521
(16)Liu,W.;Chun,E.;Thompson,A.A.;Chubukov,P.;Xu,F.;Katritch,V.;Han,G.W.;Roth,C.B.;Heitman,L.H.;Ijzerman,A P.;Cherezov,V.;Stevens,R.C.Science 2012,337(6091),232.doi:10.1126/science.1219218
(17)Boeckmann,B.;Bairoch,A.;Apweiler,R.;Blatter,M.C.;Estreicher,A.;Gasteiger,E.;Martin,M.J.;Michoud,K.;O'Donovan,C.;Phan,I.;Pilbout,S.;Schneider,M.Nucleic.Acids Res.2003,31(1),365.doi:10.1093/nar/gkg095
(18)Discovery Studio 2.5,Release 2.5;Accelrys Software Inc.:San Diego,2009.
(19)Laskowski,R.A.;MacArthur,M.W.;Moss,D.S.;Thornton,J.M.J.Appl.Cystallogr.1993,26,283.doi:10.1107/S0021889892009944
(20)Lin,K.J.;Zhu,D.J.;Leng,Y.G.;You,Q.D.Acta Phys.-Chim.Sin.2012,28(7),1783.[林克江,朱冬吉,冷勇敢,尤启冬.物理化学学报,2012,28(7),1783.] doi:10.3866/PKU.WHXB201204192
(21)Gaulton,A.;Bellis,L.J.;Bento,A.P.;Chambers,J.;Davies,M.;Hersey,A.;Light,Y.;McGlinchey,S.;Michalovich,D.;Al-Lazikani,B.;Overington,J.P.Nucleic.Acids Res.2012,40(Database issue),D1100.
(22)Xia,J.;Jin,H.;Liu,Z.;Zhang,L.;Wang,X.S.J.Chem.Inf.Model.2014,54(5),1433.doi:10.1021/ci500062f
(23)Xia,J.;Tilahun,E.L.;Reid,T.E.;Zhang,L.;Wang,X.S.Methods 2015,71,146.doi:10.1016/j.ymeth.2014.11.015
(24)Qiao,K.;Zeng,L.X.;Jin,H.W.;Liu,Z.M.;Zhang,L.R.Acta Phys.-Chim.Sin.2012,28(6),1509.[乔 康,曾凌晓,金宏威,刘振明,张亮仁.物理化学学报,2012,28(6),1509.] doi:10.3866/PKU.WHXB201203272
(25)Wolber,G.;Langer,T.J.Chem.Inf.Model.2005,45(1),160.doi:10.1021/ci049885e
(26)Schrödinger Suite 2013;Schrödinger,LLC:New York,2013.
(27)Ballesteros,J.A.;Weinstein,H.Methods Neurosci.1995,25,366.
(28)Kiesman,W.F.;Zhao,J.;Conlon,P.R.;Dowling,J.E.;Petter,R.C.;Lutterodt,F.;Jin,X.;Smits,G.;Fure,M.;Jayaraj,A.;Kim,J.;Sullivan,G.;Linden,J.J.Med.Chem.2006,49(24),7119.
(29)Giovannoni,M.P.;Vergelli,C.;Cilibrizzi,A.;Crocetti,L.;Biancalani,C.;Graziano,A.;Dal Piaz,V.;Loza,M.I.;Cadavid,M.I.;Diaz,J.L.;Gavalda,A.Bioorg.Med.Chem.2010,18(22),7890.doi:10.1016/j.bmc.2010.09.043
(30)Chang,L.C.;Kuenzel,J.V.;Mulder-Krieger,T.;Westerhout,J.;Spangenberg,T.;Brussee,J.;Ijzerman,A.P.J.Med.Chem.2007,50(4),828.doi:10.1021/jm0607956
(31)Scheiff,A.B.;Yerande,S.G.;El-Tayeb,A.;Li,W.;Inamdar,G.S.;Vasu,K.K.;Sudarsanam,V.;Muller,C.E.Bioorg.Med.Chem.2010,18(6),2195.doi:10.1016/j.bmc.2010.01.072
(32)Novellino,E.;Cosimelli,B.;Ehlardo,M.;Greco,G.;Iadanza,M.;Lavecchia,A.;Rimoli,M.G.;Sala,A.;Da Settimo,A.;Primofiore,G.;Da Settimo,F.;Taliani,S.;La Motta,C.;Klotz,K.N.;Tuscano,D.;Trincavelli,M.L.;Martini,C.J.Med.Chem.2005,48(26),8253.
(33)Mishra,C.B.;Barodia,S.K.;Prakash,A.;Kumar,J.B.S.;Luthra,P.M.Bioorg.Med.Chem.2010,18(7),2491.doi:10.1016/j.bmc.2010.02.048
(34)Moorjani,M.;Zhang,X.;Chen,Y.;Lin,E.;Rueter,J.K.;Gross,R.S.;Lanier,M.C.;Tellew,J.E.;Williams,J.P.;Lechner,S.M.;Malany,S.;Santos,M.;Ekhlassi,P.;Castro-Palomino,J.C.;Crespo,M.I.;Prat,M.;Gual,S.;Diaz,J.L.;Saunders,J.;Slee,D.H.Bioorg.Med.Chem.Lett.2008,18(4),1269.doi:10.1016/j.bmcl.2008.01.036
(35)Gillespie,R.J.;Cliffe,I.A.;Dawson,C.E.;Dourish,C.T.;Gaur,S.;Jordan,A.M.;Knight,A.R.;Lerpiniere,J.;Misra,A.;Pratt,R.M.;Roffey,J.;Stratton,G.C.;Upton,R.;Weiss,S.M.;Williamson,D.S.Bioorg.Med.Chem.Lett.2008,18(9),2924.doi:10.1016/j.bmcl.2008.03.072
(36)Gillespie,R.J.;Cliffe,I.A.;Dawson,C.E.;Dourish,C.T.;Gaur,S.;Giles,P.R.;Jordan,A.M.;Knight,A.R.;Lawrence,A.;Lerpiniere,J.;Misra,A.;Pratt,R.M.;Todd,R.S.;Upton,R.;Weiss,S.M.;Williamson,D.S.Bioorg.Med.Chem.Lett.2008,18(9),2920.doi:10.1016/j.bmcl.2008.03.076
(37)Gillespie,R.J.;Bamford,S.J.;Clay,A.;Gaur,S.;Haymes,T.;Jackson,P.S.;Jordan,A.M.;Klenke,B.;Leonardi,S.;Liu,J.;Mansell,H.L.;Ng,S.;Saadi,M.;Simmonite,H.;Stratton,G.C.;Todd,R.S.;Williamson,D.S.;Yule,I.A.Bioorg.Med.Chem.2009,17(18),6590.doi:10.1016/j.bmc.2009.07.078
(38)Silverman,L.S.;Caldwell,J.P.;Greenlee,W.J.;Kiselgof,E.;Matasi,J.J.;Tulshian,D.B.;Arik,L.;Foster,C.;Bertorelli,R.;Monopoli,A.;Ongini,E.Bioorg.Med.Chem.Lett.2007,17(6),1659.doi:10.1016/j.bmcl.2006.12.104
(39)Firooznia,F.;Cheung,A.W.;Brinkman,J.;Grimsby,J.;Gubler,M.L.;Hamid,R.;Marcopulos,N.;Ramsey,G.;Tan,J.;Wen,Y.;Sarabu,R.Bioorg.Med.Chem.Lett.2011,21(7),1933.doi:10.1016/j.bmcl.2011.02.053
(40)Cheung,A.W.;Brinkman,J.;Firooznia,F.;Flohr,A.;Grimsby,J.;Gubler,M.L.;Guertin,K.;Hamid,R.;Marcopulos,N.;Norcross,R.D.;Qi,L.;Ramsey,G.;Tan,J.;Wen,Y.;Sarabu,R.Bioorg.Med.Chem.Lett.2010,20(14),4140.doi:10.1016/j.bmcl.2010.05.056
(41)Kalla,R.V.;Elzein,E.;Perry,T.;Li,X.;Palle,V.;Varkhedkar,V.;Gimbel,A.;Maa,T.;Zeng,D.;Zablocki,J.J.Med.Chem.2006,49(12),3682.doi:10.1021/jm051268+
(42)Kim,Y.C.;Ji,X.;Melman,N.;Linden,J.;Jacobson,K.A.J.Med.Chem.2000,43(6),1165.doi:10.1021/jm990421v
(43)Stefanachi,A.;Nicolotti,O.;Leonetti,F.;Cellamare,S.;Campagna,F.;Loza,M.I.;Brea,J.M.;Mazza,F.;Gavuzzo,E.;Carotti,A.Bioorg.Med.Chem.2008,16(22),9780.doi:10.1016/j.bmc.2008.09.067
(44)Da Settimo,F.;Primofiore,G.;Taliani,S.;Marini,A.M.;La Motta,C.;Simorini,F.;Salerno,S.;Sergianni,V.;Tuccinardi,T.;Martinelli,A.;Cosimelli,B.;Greco,G.;Novellino,E.;Ciampi,O.;Trincavelli,M.L.;Martini,C.J.Med.Chem.2007,50(23),5676.doi:10.1021/jm0708376
(45)Priego,E.M.;Kuenzel,J.V.;Ijzerman,A.P.;Camarasa,M.J.;Perez-Perez,M.J.J.Med.Chem.2002,45(16),3337.doi:10.1021/jm0208469
(46)Melman,A.;Wang,B.;Joshi,B.V.;Gao,Z.G.;Castro,S.;Heller,C.L.;Kim,S.K.;Jeong,L.S.;Jacobson,K.A.Bioorg.Med.Chem.2008,16(18),8546.doi:10.1016/j.bmc.2008.08.007
(47)Baraldi,P.G.;Cacciari,B.;Moro,S.;Spalluto,G.;Pastorin,G.;Da Ros,T.;Klotz,K.N.;Varani,K.;Gessi,S.;Borea,P.A.J.Med.Chem.2002,45(4),770.doi:10.1021/jm0109614
(48)Colotta,V.;Catarzi,D.;Varano,F.;Capelli,F.;Lenzi,O.;Filacchioni,G.;Martini,C.;Trincavelli,L.;Ciampi,O.;Pugliese,A.M.;Pedata,F.;Schiesaro,A.;Morizzo,E.;Moro,S.J.Med.Chem.2007,50(17),4061.doi:10.1021/jm070123v
(49)Jaakola,V.P.;Griffith,M.T.;Hanson,M.A.;Cherezov,V.;Chien,E.Y.;Lane,J.R.;Ijzerman,A.P.;Stevens,R.C.Science 2008,322(5905),1211.doi:10.1126/science.1164772
(50)Olah,M.E.;Ren,H.;Ostrowski,J.;Jacobson,K.A.;Stiles,G.L.J.Biol.Chem.1992,267(15),10764.
(51)Rivkees,S.A.;Barbhaiya,H.;Ijzerman,A.P.J.Biol.Chem.1999,274(6),3617.doi:10.1074/jbc.274.6.3617
(52)Jaakola,V.P.;Lane,J.R.;Lin,J.Y.;Katritch,V.;Ijzerman,A.P.;Stevens,R.C.J.Biol.Chem.2010,285(17),13032.doi:10.1074/jbc.M109.096974
(53)Cheng,F.;Xu,Z.;Liu,G.;Tang,Y.Eur.J.Med.Chem.2010,45(8),3459.doi:10.1016/j.ejmech.2010.04.039
(54)Thimm,D.;Schiedel,A.C.;Sherbiny,F.F.;Hinz,S.;Hochheiser,K.;Bertarelli,D.C.;Maass,A.;Muller,C.E.Biochemistry-US 2013,52(4),726.doi:10.1021/bi3012065
(55)Ivanov,A.A.;Baskin,II.;Palyulin,V.A.;Piccagli,L.;Baraldi,P.G.;Zefirov,N.S.J.Med.Chem.2005,48(22),6813.doi:10.1021/jm049418o
(56)Gao,Z.G.;Kim,S.K.;Biadatti,T.;Chen,W.;Lee,K.;Barak,D.;Kim,S.G.;Johnson,C.R.;Jacobson,K.A.J.Med.Chem.2002,45(20),4471.doi:10.1021/jm020211+
(57)Muller,C.E.;Jacobson,K.A.BBA-Biomembranes 2011,1808(5),1290.doi:10.1016/j.bbamem.2010.12.017
Comparison of the Selectivity of Human Adenosine Receptor Antagonists Based on Structure and Pharmacophore Features
ZENG Ling-Xiao LI Xin-Ran JIN Hong-Wei LIU Zhen-Ming*ZHANG Liang-Ren*
(State Key Laboratory of Natural and Biomimetic Drugs,School of Pharmaceutical,Peking University,Beijing 100191,P.R.China)
Adenosine receptors(ARs) are crucial therapeutic targets,and selective adenosine receptor antagonists are promising for numerous therapeutic applications.In this study,three dimensional models of human adenosine A1,A2B,and A3receptors(A1AR,A2BAR,A3AR,respectively) were generated by homology modeling.In addition,pharmacophore models of the antagonists of four human adenosine receptor subtypes were developed using the LigandScout 3.12 program.Furthermore,Induced Fit Docking module of Schrödinger program was implemented to investigate receptor–ligand interactions.The results show that because of the subfamily-wide conservation of the core pocket residues,the ligand binding pockets of the three raw AR homology models are extremely similar,which poses challenges for subtype selective ligand recognition.However,the pharmacophore models of the four AR subtypes differ in pharmacophore features and spatial configuration,which are also consistent with previous site-directed mutagenesis studies.This indicates that binding site optimization is a crucial step in model generation,and the distributions for a set of pharmacophore features in ligand-based pharmacophore,including hydrogen bond acceptors,hydrogen bond donors,hydrophobic centroids,and aromatic rings,can reflect the position and direction characterization ofhydrogen bonds and hydrophobic cavities,which aid identification and characterization of binding sites.This study may provide a significant theoretical foundation for further raw model optimization in homology modeling and discovery of novel selective human adenosine receptor antagonists.
Pharmacophore; Human adenosine receptor; Antagonist; Selectivity
April 17,2015;Revised:May 25,2015;Published on Web:May 25,2015.
O641
icle]
10.3866/PKU.WHXB201505253 www.whxb.pku.edu.cn
*Corresponding authors.ZHANG Liang-Ren,Email:liangren@bjmu.edu.cn;Tel:+86-10-82802567.LIU Zhen-Ming,Email:zmliu@bjmu.edu.cn;Tel:+86-10-82805514.
The project was supported by the National Natural Science Foundation of China(21272017) and Doctoral Fund of Ministry of Education of China(20090001120049).
国家自然科学基金(21272017)和教育部博士点基金(20090001120049)资助项目
© Editorial office of Acta Physico-Chimica Sinica
