锂离子电池TiO2纳米管负极材料
2015-09-03汪倩雯杜显锋陈夕子徐友龙
汪倩雯 杜显锋,2,* 陈夕子 徐友龙,2
(1西安交通大学,电子陶瓷与器件教育部重点实验室,西安 710049;2西安交通大学国际电介质研究中心,西安 710049)
锂离子电池TiO2纳米管负极材料
汪倩雯1杜显锋1,2,*陈夕子1徐友龙1,2
(1西安交通大学,电子陶瓷与器件教育部重点实验室,西安 710049;2西安交通大学国际电介质研究中心,西安 710049)
锂离子电池负极材料二氧化钛(TiO2)由于其零应变、环境友好和高安全性近年来得到了广泛的研究,但其较低的电子电导和离子迁移率以及较低的比容量(335 mAh·g–1)限制了其应用前景.本文梳理了一种纳米结构TiO2纳米管(TNTs)的研究历程以及最近研究进展,综述了TNTs常见的几种制备方法,即水热法、阳极氧化法和模板法及其形成机理,归纳了各种制备方法的优缺点,讨论了制备过程中各项参量对制得TNTs的影响.阐述了其晶体结构与形貌对电化学性能的影响,指出晶格取向一致、管壁厚度小,纳米管开口且同向排列的TNTs具有更好的电化学性能.同时探讨了针对该材料电导性差、比容量低而进行的包括结构设计、掺杂、复合等一系列改进措施,指出与高电导率及高比容量材料复合是一种方便有效的改进措施.最后总结了各种改性方法取得的进展及存在的不足,展望了TNTs的研究趋势和发展前景.
TiO2纳米管; 负极材料; 电化学性能; 锂离子电池
Key Words:TiO2nanotube; Anode material; Electrochemical performance; Lithium ion battery
1 引 言
锂离子电池因具有高电压、高比能量、长寿命、以及环境友好等优点,获得了来自学术界和产业界的高度关注.自1990年Sony公司实现商业化以来,已成为当今世界应用最为广泛的二次电池,尤其在便携电子设备领域,占据绝对优势地位.然而,在那些需要高能量密度、高功率密度、高安全性和长寿命领域,如电动工具、电动汽车、可再生能源等,锂离子电池还不能满足需要.因此,开发具备上述特性的动力锂离子电池成为当今的研究热点.
目前,商业化锂离子电池的负极材料主要为石墨,在锂离子插入与脱嵌过程中,石墨体积极易膨胀与收缩,导致其皴裂、粉化、脱落,造成容量衰减.此外,其嵌脱锂电位(~0.1 V(vs Li/Li+))十分接近金属锂形成电位,容易导致嵌锂过程中锂枝晶形成并刺穿隔膜,引起电池内部短路乃至发生爆炸.同时,石墨电极表面形成的不导电固体电解质界面(SEI)膜以及较低的锂离子扩散系数,使得锂离子电池在高倍率性能方面表现不尽人意,这显然不能满足动力锂离子电池对大功率、高安全性的需求.因而,寻找一种新的锂离子电池负极材料,成为开发下一代锂离子电池的关键.1,2
近年来,TiO2受到研究人员的广泛关注,它在自然界储量大,对环境友好,且充放电过程中体积变化小(<3%,被称为零应变材料),嵌Li脱Li电位较高(1.4–1.8 V(vs Li/Li+)),3可以有效避免锂枝晶的形成,极大提高了锂离子电池的安全性,同时也抑制了循环过程中SEI膜的不断形成与脱落,减少容量的损失.4,5这使其成为一种新型锂离子电池负极材料.
尽管具有上述诸多优点,但TiO2电子电导差(ca 10–12–10–7S·cm–1),离子扩散系数低(ca 10–15–10–9cm2·s–1),比容量不高(最大理论比容量335 mAh·g–1),难以适应电动汽车等大功率设备的储能供电需求.解决此问题的一个有效方法是制备纳米结构TiO2.相较于微米及以上尺寸材料,纳米材料具有①能提供更短的离子电子扩散距离;②能提供更大的电极与电解液接触面积;③能更好地适应脱嵌锂过程中材料体积变化产生的应力;④能发生材料为块状结构时不能发生的新反应等优点.6作为一种典型的具有纳米结构的TiO2,TNTs可以利用水热法、阳极氧化法等方法简单快捷制备.同时研究证明,相较于其他纳米结构TiO2,TNTs具有更优的电化学性能,因而受到研究人员的广泛关注.7,8
本文对锂离子电池TNTs负极材料进行了综述,比较了水热法、阳极氧化法、模板法三种常见TNTs制备方法的优缺点,讨论了制备工艺对材料晶型与形貌的影响,总结了纳米管形貌、晶型等因素对TNTs电化学性能的影响规律,论述了目前改善TNTs的主要方法及效果,并对TNTs未来研究方向进行了展望.
2 TNTs的制备
目前,制备TNTs的方法主要有水热法、阳极氧化法和模板法.研究人员通过调整制备过程中的各种变量,调控TNTs的形貌和结构,优化制备工艺,不断改进制备方法.
2.1 水热法
水热法是Kasuga等9于1998年首先提出的,即将TiO2与高浓度NaOH溶液充分混和后转入聚四氟乙烯内衬的高压反应釜中高温反应,得到的产物用去离子水和HCl或HNO3溶液充分洗涤,干燥烧结即得TiO2纳米管.利用水热法制得的TNTs顶端开口,纳米管笔直且呈交叉排列(图1a),管壁一般为3–5层,各层间间距约为0.78 nm.10图1所示为水热法制得的TNTs透射电镜(TEM)图像11及用阳极氧化法、12高分子模板法13制得的TNTs扫描电镜(SEM)图像.

图1 水热法制得的TNTs透射电镜图像(a),11阳极氧化法(c),12高分子模板法(b)13制得的TNTs扫描电镜图像Fig.1 Transmission electron microscopy(TEM) images of TNTs prepared by hydrothermal reaction(a),11scanning election microscopy(SEM) images of TNTs prepared by electrochemical anodization(c),12and template method against polymeric nanotubes(b)13
锐钛矿、9,14金红石、15锐钛矿-金红石混合物、16锐钛矿-金红石-TiO2(II)混合物17等各种晶型的TiO2均可以用于水热法制备TNTs.起始TiO2的晶型对制得的TNTs有着直接影响:Morgan等18的研究表明,起始物为锐钛矿时,可以更加快速转化为纳米管并有更高的产量;Zhang等19发现以金红石为起始物制得的纳米管与锐钛矿制得的相比,纳米管长度明显增加;Choi等16发现,当起始物全部为金红石时,能制得管径更小、更均匀的TNTs,当采用锐钛矿和金红石混合物作为TNTs起始物时,金红石质量分数超过50%后,制得的TNTs管径明显增大;Gajovic等17研究表明,纯锐钛矿制得的TNTs与混晶(锐钛矿-金红石-TiO2(II)混合物)制得的TNTs相比,其晶相转变温度更高,前者制得的TNTs在300°C才开始转变为锐钛矿相,而后者在80°C即有锐钛矿相出现;以金红石纳米粉体为起始物时,随着粉体粒径的减小,制得的TNTs长度增加、孔径减小、均匀性增加,当起始物粒径大于100 nm时,水热反应转化不完全,产物中仍有许多球形颗粒存在.19
此外,水热反应的温度、时长、NaOH浓度也会影响产物的形貌:水热法反应温度一般在100–150°C,温度过低(<70°C),产物为片状,温度过高(>170°C),产物为纳米线;20–22反应时间一般为20–120 h,时间过长(12 d),产物为纳米线,时间过短(5 h),产物为许多纳米管团聚体而没有单根纳米管;17,19当NaOH浓度低于5 mol·L–1或高于20 mol·L–1时,很难生成纳米管,当NaOH浓度为5–10 mol·L–1时,生成的纳米管结构和产量最理想.23
作为最早发明水热法的研究人员,Kasuga等9认为纳米管是在酸洗水洗过程中形成的.Sun24和Ma25等采用类似的方法来合成纳米管,也认为水洗有助于纳米管的形成.这种观点认为,在水热反应的初始阶段,TiO2与NaOH反应形成纤铁矿型钛酸钠,在随后的酸洗和水洗过程中,H+取代Na+,同时卷曲成纳米管.26
但Du等27采用同样水热过程于130°C不经过水洗和酸洗即可得到纳米管,认为纳米管是在碱处理过程中形成的.Wang28、Yao29和Nian26等随后的研究也支持这一观点.他们认为,水热合成时,在高压、高温和强碱作用下,TiO2沿晶面被剥落成薄片,在片的两面有许多不饱和悬挂键.随着水热反应的进行,不饱和悬挂键数量增多,使得薄片表面活性增强,为了减少悬挂键数量,降低体系能量,薄片开始以(010)晶面为轴卷曲成管状结构.进一步延长水热反应时间,纳米管可以通过溶解长大机理而增长.后续的清洗对产物形貌无明显影响,30但其减少了纳米管壁中的层间水,提高了材料的比表面积.31
Kukovecz等32则提出了一种新的纳米管形成机制,他们认为纳米管不是由片状钛酸盐卷曲形成,而是少量的原料从锐钛矿微晶上剥离下来,这些微晶重结晶成片状钛酸盐,然后卷曲成单螺旋,多螺旋或葱状截面的纳米环,大多数原料以这些纳米环为晶种,通过晶体定向生长机制生长为纳米管.不少研究者认为Kukovecz的模型是目前最有可能的纳米管生长机制.31,33,34
目前关于纳米管的成分也存在着一定的争议,一部分人认为纳米管的组分为H2Ti3O7,10,27,28一部分认为是HTiO·HO,35一部分人认为是HTiO(OH),33也有人认为是HxTi2−x/4x/4O4.34根据XRD、SAED、 HRTEM等测试结果的分析,更多研究人员倾向于接受纳米管的组分为H2Ti3O7这一观点.36
水热法制得的TNTs晶型不完整,晶体微粒之间有凝聚现象,纳米管稳定性差,加热、酸或其他化学试剂处理都可能会造成其晶型再一次发生转变.8但与模板法,阳极氧化法相比,水热法简单易行,成本低,产率高,仍是一种被广泛采用的TNTs制备方法.
2.2 阳极氧化法
阳极氧化法是将钛和惰性电极组成两电极体系浸润在含氟电解液体系中,在恒定电位下将钛板进行阳极氧化,在钛板上形成TNTs阵列.采用电化学阳极氧化方法可以制得取向一致、顶端开口的TNTs阵列(如图1c),且操作简单,过程可控.
阳极氧化法中,阳极氧化电压与时间、电解液组成、温度与浓度都会影响TNTs的形貌及物化特性.
对于组成相同的电解液,TNTs的形貌主要由阳极氧化电压决定,而电解液温度和浓度仅会影响TNTs形成所需要时间.当氧化电压大于一定值后,钛箔表面不再出现管状结构,取而代之的是疏松海绵状结构.37
调配不同组分的电解液是调控阳极氧化法所得TNTs尺寸的一种常用手段.按照电解液组分分类,TNTs的发展可分为三代:38第一代电解液为HF的酸性较强水溶液(pH < 3),制得的TNTs长度一般小于500 nm;第二代电解液普遍采用弱酸性的氟化物水溶液,TNTs长度增至6 µm;第三代使用酸性更弱的有机电解液,可生长出长达500 µm的TNTs阵列.目前使用较广泛的是含水和NH4F的乙二醇有机电解液.
电解液组分不仅能调控TNTs的长度,也影响着纳米管的其他性能:Macak等38研究表明,通过配制高粘度的甘油电解液,可以有效抑制钛板表面的局部浓度起伏,得到均匀分布的TNTs阵列;Pervez等39发现,通过在电解液中加入乳酸,可以显著提高TNTs的生长速度,同时增强TNTs在钛箔上的附着力;John等40在电解液中加入离子液体1-丁基-3-甲基咪唑四氟硼酸盐(BMIM-BF4),得到了双层同轴纳米管结构;Li等41利用BMIM-BF4做乙二醇有机电解液的氟化前驱体,提高有机电解液的导电性,减少TNTs中的缺陷.目前,通过调控电解液组分来调控TNTs形貌仍然是不少研究人员研究的重点.
2.3 模板法
模板法是以适宜尺寸和结构的模板为主体,利用物理化学方法在其上合成TiO2,然后去除模板,得到TNTs.目前较常采用的模板有多孔氧化铝(PAA)模板、多孔高分子模板、介孔分子筛等.模板法制得的TNTs结构与尺寸和模板以及反应过程有关.PAA模板由于制备方法简单,目前受到较多研究者的青睐.以PAA为模板制得的纳米管取向一致,管壁笔直.该方法制得的TNTs管径由PAA孔径决定.Lakshmi等42研究发现,通过延长PPA模板在胶体溶液中浸润时间,可以获得管壁更厚、管子更长的TNTs.
高分子纳米管(PNT)模板在较高温度下可以很容易地被去除,同时高温处理也使表面的TiO2结晶,也是一种较常采用的模板.此种方法制得的TNTs没有统一取向,且纳米管弯曲封口,如图1b所示.13
模板法制备TNTs能够较为方便地控制管子的长度、管壁的厚度以及管子的直径,但此种方法难以获得内径较小的TNTs,且制备工艺复杂.另一方面,在去除模板的过程中,纳米材料的表面甚至是结构极易受到损坏.
同制备方法所得TNTs各有优劣,其特征比较见表1.

表1 三种典型TNTs制备方法的比较Table 1 Comparison of three typical synthesis methods of TNTs
3 影响TNTs电化学性能的因素
3.1 TNTs晶型
目前,锂离子TiO2负极材料研究较多的主要是锐钛矿、金红石、TiO2(B)以及无定性TiO2.一般而言,TiO2的脱嵌锂可以表示为式(1),其中x为单位TiO2中插入的锂离子摩尔数量.

研究表明:锐钛矿是最具电化学活性的TiO,43–452当x < 0.5时,(1)式反应是完全可逆的;46而当x >0.5时,锂离子几乎不能在晶格中迁移,嵌锂反应仅能在小于4 nm深的表面进行.47因此,一般认为块状锐钛矿嵌锂反应的产物为Li0.5TiO2,其理论比容量为167 mAh·g–1.但随着材料尺寸的减小,比容量随之增加.有研究者认为,当材料尺寸在7 nm以下时,嵌锂反应在x=1时仍能可逆进行,可以快速地进行脱嵌锂反应,理论容量可以达到335 mAh·g–1.48–50
尽管在体相金红石中锂离子的脱嵌较难发生,但在高温(120°C)下,金红石的电化学性能得到显著提高,51因而可以作为高温锂离子电池的理想负极材料.当金红石尺寸降到纳米量级时,(1)式在x=0.5时也能可逆进行,使其比容量达到167 mAh·g–1.52,53同时,纳米金红石也具有优越的低温性能.54
此外,考虑到金红石相较于其他几种TiO2热力学最为稳定,在自然界存在最广泛,因而也不失为一种良好的锂离子电池负极材料.
与锐钛矿比较,金红石中离子迁移占主导的嵌锂机制不同,TiO2(B)嵌锂反应主要是伪电容的感应电流过程,55可以快速地响应大倍率充放电.但其晶型为亚稳态,易转化为锐钛矿相,或进而转化为金红石相.另一方面,TiO2(B)与金红石、锐钛矿相比结构疏松,密度小,使材料的体积比容量下降.7
无定型态TNTs近年来也渐渐受到了更多的关注.研究普遍表明无定型结构的TNTs具有比锐钛矿更高的比容量,尤其是在大电流密度下.这可能是因为无定型TNTs具有更好的离子电导.56,57但无定型TNTs较差的电子电导使其在循环过程中容量损失严重(尤其是在循环前100–200圈),无法像结晶性良好的TNTs一样拥有良好的容量保持率.因而,改善无定型TNTs电子电导,提高其循环稳定性,成为了无定型态TNTs研究的重点.同时,首圈库伦效率低下也是无定型态TNTs锂离子电池负极材料存在的一个主要问题.在首圈充放电后,无定型态TNTs有高达50%的不可逆容量,而锐钛矿相TNTs中这一值仅为5%.4,56目前普遍认为造成这一现象的原因有两个:一是由于无定型TNTs中存在的结构缺陷及化学缺陷充当了Li+陷阱,在充电过程中发生了不可逆的嵌锂反应;另一个原因是在放电过程中,Li+和吸附在TNTs表面的水分子发生不可逆反应(H2O + xLi++ xe–⇌ LiOH + 1/2H2).4,58如何减少无定型TNTs的不可逆容量也成为了一些科研人员的研究重点.其中,Ryu等56通过采用有机体系阳极氧化法,排除水的影响,将无定型TNTs的首圈容量损失降低到35%.
另一方面,TNTs晶格取向也会影响材料电化学性能.对金红石相TNTs而言,C轴的电子传导明显优于其他方向,因而C轴生长的TNTs具有更好的电化学性能.59对锐钛矿而言,(001)面具有高于其他晶面的能量密度,通过控制晶粒生长,使(001)更多地暴露在表面,可以提高脱嵌锂速度,提升库伦效率和循环稳定性.60,61其次,与非取向的TNTs相比,晶格取向一致的TNTs晶界减少,材料的导电性得到改善.62
阳极氧化法、模板法制得的TNTs通常为无定型结构,一般需进行热处理形成结晶TNTs.在400°C进行热处理时,材料逐渐转换为锐钛矿相;在600°C以上进行热处理时,材料逐渐转化为金红石相.41但值得注意的是,热处理过程中往往伴随纳米管结构的坍塌并逐渐转化为纳米带.63对于阳极氧化法制得的TNTs,热处理还会使其内外管壁结构分离,阻碍电子在纳米管中的传导,从而劣化循环性能.64,65为了避免这种结构改变造成的不利影响,同时考虑到无定型TNTs较其他晶相具有更高的比容量,不少研究人员选择舍去热处理这一步骤,直接利用无定型结构的TNTs作为负极材料.66,67
3.2 TNTs的形貌
研究表明,TNTs的电化学性能与其形貌息息相关,纳米管的直径、管长、管壁厚度、排列方式等因素都会影响TNTs的性能.
材料的尺寸影响着材料的物理化学性能,尤其当尺寸小到纳米尺度,材料可以发生某些块状时不会发生的新反应.例如,对块状锐钛矿而言,(1)式中的x=0.5,材料的理论容量为167 mAh·g–1,但当材料尺寸小到7 nm以下时,嵌锂反应在x=1时仍能可逆进行,理论容量可达335 mAh·g–1.48–50Panda等68制备了一系列不同管壁厚度的TNTs,发现当TNTs的管壁厚度减少到5 nm时,材料的质量比容量为330 mAh·g–1,而当TNTs的管壁厚度达到40 nm时,容量减少至170 mAh·g–1.不仅管壁过厚会影响TNTs的性能,纳米管的长度也不能过长.Gonzalez等69研究表明,随着TNTs纳米管长度的增加,材料的导电性将显著下降.
但另一方面,纳米管尺寸的减小也会给材料带来负面影响.Pervez等39发现TNTs纳米管长度越短,直径越小,材料的面积比容量就越小.Freitas等70则发现,随着纳米管直径的减小,材料充放电循环中的容量衰减与晶格应力均随之增加.
此外,TNTs的排列方式也会影响材料的电化学性能.TNTs在集流体上的排列大致有以下三种形式:①随意无规则排列;②取向一致,纳米管封口;③取向一致,纳米管不封口(图2).Han等71研究表明,同等情况下,取向一致,不封口的TNTs表现出更高的比容量和循环稳定性.这是因为取向一致的排列方式使其与集流体的接触电阻显著减小,同时开口的纳米管结构为电解液和活性材料提供更大的接触面积.

图2 TNTs排列示意图Fig.2 Schematic illustration of TNTs
如何调控TNTs的各项尺寸及排列方式,使其总体性能最优化,仍然是目前不少研究人员研究的重点.
3.3 TNTs的表面性质
TNTs的形貌和晶型对材料的比容量有直接的影响.但除了比容量外,材料的首圈库伦效率也是衡量其电化学性能的一个重要指标.过低的库伦效率会造成活性物质的浪费,使电极能量密度下降.而表面化学性质是决定材料首圈库伦效率的主要因素,72–75在TNTs中,纳米管形貌使活性物质与电解液有更大的接触面积,材料的表面化学特性对库伦效率的影响显得更为突出.
材料首圈容量损失只要来源于以下几个方面:(1) 材料表面存在的结构缺陷及化学缺陷充当了Li+陷阱,在充电过程中发生了不可逆的嵌锂反应;(2) 在放电过程中,Li+和吸附在材料表面的水分子发生不可逆反应(H2O + xLi++ xe–=LiOH + 1/2H2).4,58(3) TiO2表面具有大量的O-H基团和结合水,电解液和这些基团发生不可逆副反应(如H2O + LiPF6=LiF + F3PO + 2HF,F3PO + 3ROH =(RO)3PO + 3HF),造成了首圈库伦效率的低下.76,77
因而,通过调控TNTs表面特性,减少表面化学缺陷和结构缺陷,减少结合水和O−H数目,可以显著改善材料首圈库伦效率.Brutti等78用C2H5OLi对TNTs进行预处理,反应Ti-O-H + C2H5OLi=Ti-O-Li + C2H5OH显著减少了材料表面的O-H数目,使材料首圈库伦效率上升近20%.通过热处理,79在无水电解液体系中制备TNTs,56也可以显著减少表面结合水和O-H数目,提升材料首圈库伦效率.同时,通过优化制备工艺,41在材料表面进行碳层或金属氧化物包覆,80,81减少其表面化学和结构缺陷,也是常用的提升首圈库伦效率的方法.
4 TNTs改性
目前,制约TNTs作为锂离子电池负极材料应用的主要因素是其较低的比容量和较低的电导率.针对这些问题,研究人员采用了TNTs结构设计、TNTs表面负载、杂质离子掺杂、TNTs与其他材料混合等方法对TNTs进行改性,从而制备①比表面积大,②导电性好,③抗凝聚,④比容量高的材料.
4.1 设计结构
微观结构对材料的电化学性能有着显著的影响.TNTs的纳米管结构提供了大比表面积、短电子、锂离子传输距离,使材料与块状TiO2相比,性能得到极大提升.研究人员通过对TNTs进行结构设计,使其纳米管排列更为疏松,与电解液接触面积更大,导电性更好,从而进一步提升材料的电化学性能.
4.1.1 在多孔基底上生长TNTs
锂离子电池TNTs负极常见制备方法多采用阳极氧化法在平整钛箔上直接生长TNTs,或是将制得的TNTs与添加剂混合涂覆在集流体上.近年来,研究人员通过设计一些新颖结构的基底,将TNTs生长在这些基底上,可以大幅增大TNTs的生长面积,使活性物质与电解液具有更大的接触面积.另一方面,这些基底往往具有优于TiO2的导电性,可以较好地改善材料导电性,提升材料大电流充放电性能.
Bi等82采用电子束溶解技术(EBM)制备泡沫钛(Ti),并在其上用电化学方法生长TNTs.疏松多孔的泡沫Ti不仅为活性材料和集流体之间提供了良好的电导,而且相比于普通Ti箔,它提供了更大的场所生长TNTs,生长于其上的TNTs也更为疏松(图3a).因此,与普通Ti上生长TNTs相比,这种结构表现出更为优越的电化学性能,比容量可以达到前者的两倍.

图3 生长在Ti泡沫(a)、82Ti网(b)、83碳纳米纤维网(c)84上的TNTs示意图Fig.3 Schematics of the TNTs grown on the surface of Ti foam(a),82Ti mesh(b),83and C nanofiber(c)84
Zhang等83通过阳极氧化法在钛网上制备三维TNTs阵列(图3b),这种材料可以直接用作电极而不需要集流体与添加剂,起骨架作用的薄钛网可以保持良好的导电性.特殊三维结构为活性物质和电解液提供了更大的接触面积,更短的锂离子传输距离,从而使材料具有更好的电化学性能.随着阳极氧化时间的增长,样品电化学性能显著提升,在50 µA·cm–2的电流密度下恒流充放电循环100圈,材料的面积比容量为1745.5 µAh·cm–2.另一方面,用钛网取代钛薄片提高了钛的利用率,节省了原材料并降低了成本.
对基底的改良不仅仅局限于钛.Zhao等84以交叉排列的碳纳米纤维为基底,在其上生长TNTs,得到了一种三维多孔的纳米结构,如图3c所示,在1C、2C、5C倍率下,材料放电比容量分别为306、249、214 mAh·g–1,碳纤维良好的导电性使材料拥有了优异的倍率性能,同时,材料也表现出良好的循环稳定性,即使是在30C大倍率循环下,电池循环1000圈儿依然没有容量衰减.
4.1.2 在TNTs中引入其他结构
TNTs的电化学性能与其尺寸息息相关.然而在调控TNTs尺寸时发现,材料各项性能的优化无法得到统一.尽管减小TNTs的尺寸可以增大材料的比容量,但材料在循环过程中易于凝聚,导致循环过程中容量衰减严重;增大TNTs长度可以显著提高材料的面积比容量,但质量比容量下降,导电性变差,材料大电流充放电性能变差.据此,研究人员通过在TNTs材料中引入其他结构,同时优化各项性能.
Chen等85制备了表面覆盖TNTs的中空微米球(图4),微米球直径约为400 nm,表面TNTs长200 nm,直径30 nm,管壁厚度5 nm,将中空微米球与TNTs两种结构融合,提高了材料的导电率,抑制了材料在充放电循环中的凝聚,从而提高了材料的比容量和循环稳定性.1C(337 mA·g–1)倍率充放电500圈后,比容量仍然维持在150 mAh·g–1,8C时,比容量仍有90 mAh·g–1.
生长在集流体上的TNTs阵列越厚,面积比容量越大,但导电性也随之下降.Song等86设计一种CTiO2树即在碳纳米纤维上生长TNTs,将较短的TNTs与较长的碳纤维在材料中统一,解决了TNTs中高面积比容量与高电导率不能兼得的问题.与直接生长在集流体上的TNTs相比,这种结构的TNTs有10倍于前者的面积比容量.在1000 mA·g–1电流密度下,放电比容量达150 mAh ·g–1.
对TNTs进行结构设计可以在一定程度上改善材料的导电率,提高比容量,改善材料电化学性能.但工艺相对复杂(如在中空微米球上生长TNTs,在碳纤维上生长TNTs),因而,不少研究人员采用掺杂这种工艺相对简单的方法来提升TNTs的电化学性能.
4.2 掺 杂
TiO2半导体性质使其电导率较低,不利于电子的快速扩散,大电流下极化严重.通过对TNTs进行掺杂,可以引入缺陷提高电导率,提高材料的电化学性能.

图4 表面生长TNTs的中空TiO2微米球TEM图像85Fig.4 TEM image of TiO2hollow microspheres with the shell consisting of nanotubes85
4.2.1 Ti3+掺杂
Ti3+掺杂在晶格中引入了氧空位,能有效提高TNTs的电子电导,从而提升锂离子电池的倍率性能,同时,相比于在TNTs中引入其他元素,这种掺杂工艺相对简单、易于制备,是改善TNTs电化学性能的常用方法之一.目前的报导中对TNTs进行Li+掺杂的主要手段有:①在还原气氛下进行高温处理,②电化学还原.
在还原气氛(H2,87,88Ar,89真空62)下高温退火被广泛用于还原TNTs,达到掺杂Ti3+目的,这种方法操作简单,还原率高.在真空中还原,Ti4+的还原率可达31.7%,62制得的材料应用于锂离子电池、超级电容器,其性能都有较大的提升.但这种方法也存在着耗时长(大于10 h),涉及高温、高压、可燃气体(如H2)等危险因素的问题.
与高温还原法相比,电化学还原法更为快速、便捷、安全.Guo等90在电解池中搭建两电极体系,以电化学还原法还原TNTs,这种方法耗时短,更安全,但Ti4+还原率较低,仅为1%.Li等91采用Ti、Pt、Ag/AgCl三电极体系还原TNTs,将Ti的还原率提升至22%.大量的氧空位显著提高了材料的电导率,改善了材料的循环稳定性,将材料应用于锂离子电池负极,在100 mV·s–1的扫描速率下,循环2000圈后容量损失仅为1.9%.
考虑到TNTs表面的Ti3+不稳定,易被空气中的氧氧化,92Zhang等93制备了TiO2-δ-La 复合物纳米管.利用Ti-O-La化学键固定在水热法制备中产生的Ti3+缺陷.这种锂离子电池负极材料性能得到了明显改善,在20C倍率下可逆容量仍有142 mAh·g–1,以10C倍率循环1000圈,仍能保持87%的可逆容量.
4.2.2 掺杂其他元素
除Ti3+掺杂以外,研究人员对TNTs也进行了C、N等其他元素的掺杂.
将TNTs与掺杂源进行高温处理是一种常用的掺杂手段.Kim等12将制得的TNTs置于C2H2/N2(20%(φ) C2H2)混合气氛中,在500°C下热处理,C原子取代了TiO2晶格中部分O原子,形成C掺杂,提高了TNTs的导电性,从而改善了其电化学性能,在50 mA·g–1电流密度下循环30圈后,纯TNTs的比容量仅为140 mAh·g–1,而掺C 的TNTs为180 mAh·g–1.Li等94将TNTs/石墨烯混合物与尿素混合在400°C下进行热处理,在TNTs中掺入N元素,提高了TNTs导电性,有效提升了材料的大电流充放电性能,在5 A·g–1的电流密度下循环180圈后,材料的可逆容量仍有90 mAh·g–1.
TNTs高温掺杂常常涉及高温、高压、可燃气体等危险因素,且掺杂多发生在TNTs的表面,因而,部分研究人员选择在纳米管形成之前完成对TiO2的掺杂.Xu等95首先制备掺杂C的TiO2粉末,然后以其为前驱体利用水热法制备C掺杂的TNTs,将C的掺杂量提高至2%,在50 mA·g–1的电流密度下循环30圈后,质量比容量为211 mAh·g–1,明显优于利用高温掺杂碳的TNTs.Kyeremateng等96采用电子共溅射法制备Ti-Sn合金薄膜,然后利用阳极氧化法得到Ti1–xSnxO2纳米管,Sn取代了TNTs中部分Ti,当x >0.05时,材料从锐钛矿相向金红石相转变,金红石结构更有利于锂离子的扩散,51,97Ti1–xSnxO2纳米管的嵌锂反应比纯TNTs快40倍.23同时,这种方法还被用于Fe、Sb、Nb等元素在TNTs中的掺杂.
用于TNTs掺杂的元素和方法不仅限于上述.利用浸渍法、98离子交换法、99,100在阳极氧化的电解液中加入掺杂离子,101–103研究人员成功地在TNTs中掺杂了Fe、104F、105Cu、106La、107Ni、108Mn、109Co、104,105Nb110等元素.但这些元素的掺杂对TNTs的导电性及比容量并无明显的改善,而是调整了TNTs的能隙,调控其响应光谱和光催化速率,多用于太阳能电池及光催化.
通过对TNTs进行掺杂,能在一定程度上改善材料的电化学性能,但掺杂在很大程度上并没有改变TNTs的分子结构和脱嵌锂反应原理,并不能大幅度提升材料比容量,同时,纳米材料易凝聚的问题仍然没有得到解决.据此,一些研究人员考虑在TNTs表面负载高电导率和或高比容量的材料来改善TNTs的电化学性能.
4.3 TNTs表面负载其他材料
在TNTs表面负载其他材料的目的大致有以下四点:①TiO2循环稳定性好,但比容量小,而许多比容量大(如Si比容量3580 mAh·g–1)的材料循环性能差,通过在TNTs表面负载这些材料,可以得到兼具两者优点的复合材料;②负载导电性高的材料,增强材料导电性;③TNTs易于凝聚,通过表层的负载,可以减少纳米管之间的凝聚现象,提高循环稳定性;④提高材料密度,从而提高面积比容量、体积比容量.同时,一个结构完整的负载层可以显著减少活性物质与电解液接触面的结构缺陷,从而减少不可以的嵌锂反应,提高材料首圈库伦效率.111TNTs表面负载方法多种多样,目前报导的方法主要有:化学气相沉积、光沉积、电化学沉积、原子层沉积、溶胶-凝胶法、磁控溅射等.
4.3.1 负载高比容量材料
TiO2尽管价格低廉,来源广泛,对环境友好,循环性能好,但其较低的容量(167–335 mAh·g–1)限制了它的应用.同时,一些材料具有很高的理论容量,但其在脱嵌锂过程中体积变化剧烈,使得SEI膜循环中不断地脱落与重建,从而造成活性物质与电解液的损失,电池容量衰减过快.通过在TNTs表面负载这些高比容的材料,可以得到比容量较高的复合材料,同时,TNTs作为基底,有良好的循环稳定性,有效遏制了比容量在循环过程中的衰减.
如3.2节中所述,相较于TNTs的其他排列方式,顶端开口、纳米管取向一致的TNTs阵列具有更好的电化学性能,因而不少研究人员选择此种TNTs阵列作为负载其他材料的载体,为了防止负载材料过厚而破坏TNTs的微观结构,常采用沉积、溅射等易于控制负载材料厚度的方法.Ivanov等112利用直流磁控溅射,Brumbarov等113采用等离子化学气相沉积(PECVD),先后成功地在TNTs阵列上涂覆一层Si薄膜.Guan等114则采用电化学沉积的方法,在TNTs阵列的顶端负载MoO3;Fan等115以紫外光激发的光沉积方法在TNTs上负载Co3O4.与未负载的TNTs相比,负载后材料的面积比容量,质量比容量均得到显著增加.例如,通过负载MoO3薄膜,材料的面积比容量最大可增长8倍之多.同时,相比微米级材料,纳米级薄膜在体积变化时会优先沿着薄膜表面方向,从而有效地避免了SEI膜的脱落和重建,获得更为优越的循环稳定性.116
但是,选择纳米管取向一致的TNTs阵列作为载体时,负载材料通常仅能涂覆在纳米管的端面及管壁上侧,可供涂覆的面积小,且不宜过厚,否则会破换纳米管形貌.然而,在一定范围内,负载的高比容量材料越多,复合材料的比容量越大,如图5所示因而,不少研究人员选择随机排列的TNTs作为载体,从而使纳米管外壁上材料的负载量增加,提高复合材料的比容量.Xu等13用溶剂热法在TNTs外壁附着MoS2纳米薄片,由于纳米管的外壁提供了远大于端面的负载面积,从而可以负载更多的其他材料,更显著地提高材料的比容量,该材料在100 mA·g–1电流密度下循环20圈比容量可达600 mAh·g–1(TNTs的最大理论容量为335 mAh·g–1).
此外,部分研究人员选择在TNTs管壁内侧负载其他材料.Wu等117利用电化学方法,直接在TNTs阵列中生长同轴的SnO2纳米管.Jeun等118则以聚丙烯腈纳米纤维为模板,利用原子层沉积技术(ALD)先后进行两次沉积,制备了以TiO2为外层、SnO2为内层的双层纳米管.这种方法尽管负载工艺较为复杂,但也有其可取之处:与将材料负载在管端相比,这种方法能提供更大的负载面积;与将材料负载在外壁相比,这种方法能够更好地抑制高比容量材料脱嵌锂过程中的体积变化;另外,在TNTs内壁负载其他材料,不会增大材料的总体积,提高材料比容量的同时大大提高了材料的振实密度,使材料的体积比容量得到大幅提升.
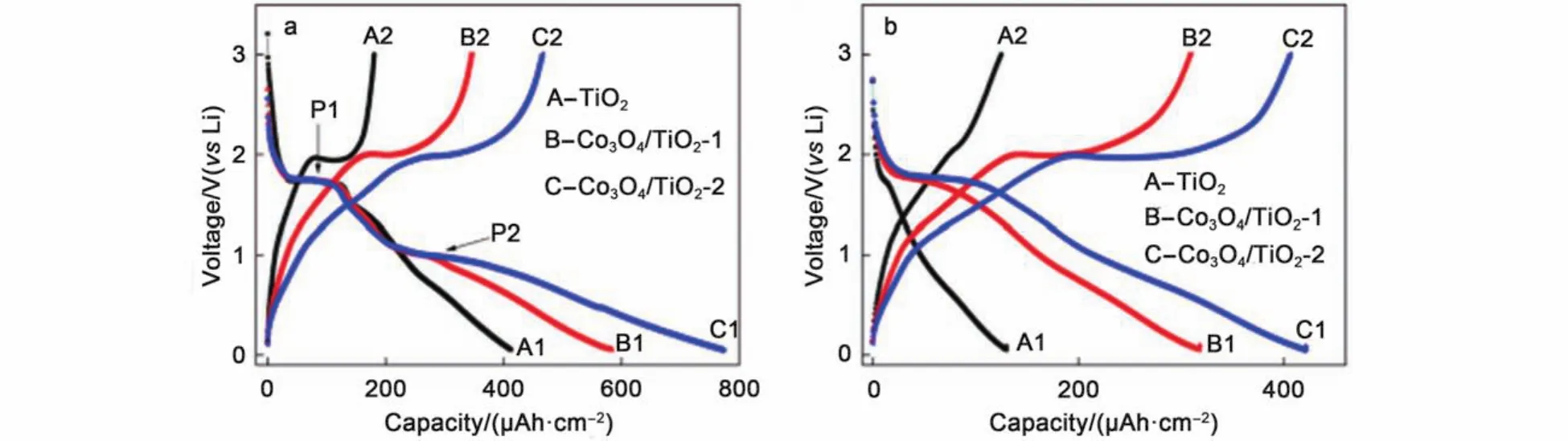
图5 纯TNTs(A)、Co3O4/TiO2-1(Co3O4负载量:0.12 mg·cm–2)(B),Co3O4/TiO2-2(Co3O4负载量:0.25 mg·cm–2)(C)作为半电池的负极材料在充放电循环第1圈(a)和第10圈(b)的电压–容量关系119Fig.5 Voltage· versus capacity curves of assembled half-cells by usin·g bare TiO2(A),as-synthesized Co3O4/TiO2-1(Co3O4loading:0.12 mgcm–2)(B),and Co3O4/TiO2-2(Co3O4loading:0.25 mgcm–2)(C) electrodes as anodes in(a) the 1st cycle,and(b) the 10th cycles119
4.3.2 负载高电导率材料
通过减小TiO2粒径,制备纳米尺度的TNTs,可有效缩短材料中锂离子的扩散距离,提高材料导电性,但TiO2本身的半导体性质仍然抑制了TNTs的导电性,使TNTs在大电流密度下极化严重.因而,研究人员通过在TNTs表面负载一层具有高电导率的材料,提高材料的整体导电性,从而提高材料的电化学性能.
碳来源广泛、成本较低、导电性好、制备工艺较为简单,因而成为一种被广泛应用的TNTs表面负载材料.Kang等120利用溶胶凝胶法在TNTs表面包覆聚乙烯吡咯烷酮(PVP)薄膜,然后热处理在TNTs表面涂覆碳薄膜;Park等121则进一步简化了工艺,直接在水热法制备TNTs时加入葡萄糖,得到了表面负载碳薄膜的TNTs,负载在TNTs表面的碳薄膜提高了TNTs的导电性,从而提升了TNTs的大电流充放电性能以及容量保持率.在7500 mA·g–1的电流密度下充放电循环,材料的比容量仍有150 mAh·g–1,而纯TNTs中这一值仅为50 mAh·g–1,同时碳薄膜的负载也有效地抑制了水热法及后续退火处理中TNTs的凝聚,有利于抑制循环中比容量的衰减;Bresser等122在水热法制得的TNTs上吸附丙烯腈、丙烯酰胺、多巴胺的共聚物,然后进行热分解处理,获得表面负载碳薄膜的TNTs,碳薄膜的负载使材料呈现出良好的循环稳定性和大电流充放电性能,平均每圈比容量衰减为0.02%,在10C、15C的倍率下,比容量分别为130、110 mAh ·g–1.
导电聚合物也可用于TNTs的改性.通过将TNTs在双三氟甲基磺酰亚胺锂(LiTFSI)和二甲基丙烯酸酯基聚乙二醇(MA-PEG)的混合溶液中进行电聚合,Plylahan等123在TNTs表面负载了聚合物胶体,与碳薄膜不同的是,这种含有LiTFSI离子的胶状聚合物具有良好的离子电导,在增强材料导电性的同时不会阻碍TNTs与锂离子的接触,制得的材料电化学性能较负载前显著增加,在1/10C,1/5C,1C的电流密度下容量分别提升25%、25%、45%.
除了碳膜及聚合物,金属因其良好的导电性也被用于TNTs负载,在各种金属中,银电导率最高,被广泛采用.He等124采用传统的银镜反应,在TNTs表面负载Ag层,Ag在TNTs中形成导电网络,其良好的导电性提升了材料大电流下的比容量和循环稳定性,在600 mA·g–1电流密度下恒流充放电循环600圈后,Ag/TNTs比容量衰减为6.99%,而纯TNTs衰减为16.57%,这种方法操作简单,但Ag颗粒较大,且不均匀;Fang等125将阳极氧化法制得的TNTs热处理,然后在2%硝酸银溶液中浸泡5 d,然后在空气中以10°C·min–1的速率升温至450°C,保温5 h后自然冷却,得到表面负载纳米Ag颗粒的TNTs;Guan等114在含AgNO3的电解液中,利用电化学沉积法,在TNTs上负载纳米Ag颗粒,金属Ag的负载提高了材料的导电率,使样品的循环稳定性得到提升,容量保持率比纯TNTs高出10% .
尽管导电性略逊于Ag,但金属Sn可进行可逆的脱嵌锂反应,具有较高的比容量(992 mAh·g–1),因而也被广泛用于TNTs的涂覆,以提高复合材料的比容量:Kim等126通过对TNTs和SnCl4混合溶液中的SnCl4进行热分解反应,清洗干燥后得到表面负载金属Sn的TNTs;Kim等12利用射频磁控溅射法在TNTs表面涂覆Sn薄膜,简化了实验步骤,提高了Sn薄膜的洁净度和可控度,并利用后续热处理使Sn重新融化,从而在TNTs的内外表面形成均匀的Sn薄膜,提高了薄膜的覆盖面积和均匀性.Sn的负载提供了稳定的锂离子快速传输通道,提高了材料的大电流充放电性能,在4000 mA·g–1电流密度下(5 min完成一次充放电循环),材料的比容量仍有176 mAh·g–1,能量密度为317 mAh·cm–3,是同等条件下纯TNTs能量密度的3.5倍.同时,金属Sn薄层包覆也显著减少了活性物质与电解液接触面的结构缺陷,减少了不可逆嵌锂反应.在100 µA·cm–2电流密度下,材料首圈库伦效率由39%提升至78%.111
尽管在TNTs表面负载其他材料可以显著改善样品的导电性、比容量、大电流充放电性能、循环稳定性等指标,但其制备工艺大多相对复杂,成本高,耗时长,而且,当表面负载的薄膜过厚时,Li+嵌入/脱出TNTs过程中受到负载薄膜的阻碍,样品的比容量反而会下降.122,123
4.4 TNTs与其他材料复合
与表面负载其他材料相比,制备TNTs与其他材料的混合物工艺相对简单,对掺入其中的材料的量没有限制,可以随意调整样品中TNTs的比例.除增强材料的导电性和/或比容量外,加入的材料在TNTs中间还可以起到支撑骨架的作用,防止TNTs的凝聚,能有效地提升样品的循环稳定性,因而受到研究人员的关注.
4.4.1 TNTs与碳纳米材料复合
近年来,新兴碳纳米材料引起了广泛的关注,研究人员对TiO2与碳纳米材料的混合材料进行了研究,取得了一定的进展:Kang等127采用机械混合的方法,制备了碳纳米管(CNT)和TNTs(TNT)的混合物.CNT能够形成三维纳米网络,增强材料的电子电导,促进电解液和活性材料间Li+的扩散,同时,随着复合材料中CNT的增加,材料的导电性增强,CNT和TNT 中间的协同作用增强,材料的电化学性能也得到提升,当CNT质量分数为0、10%、20%时,样品循环100圈后的比容量分别为130、160、180 mAh·g–1.
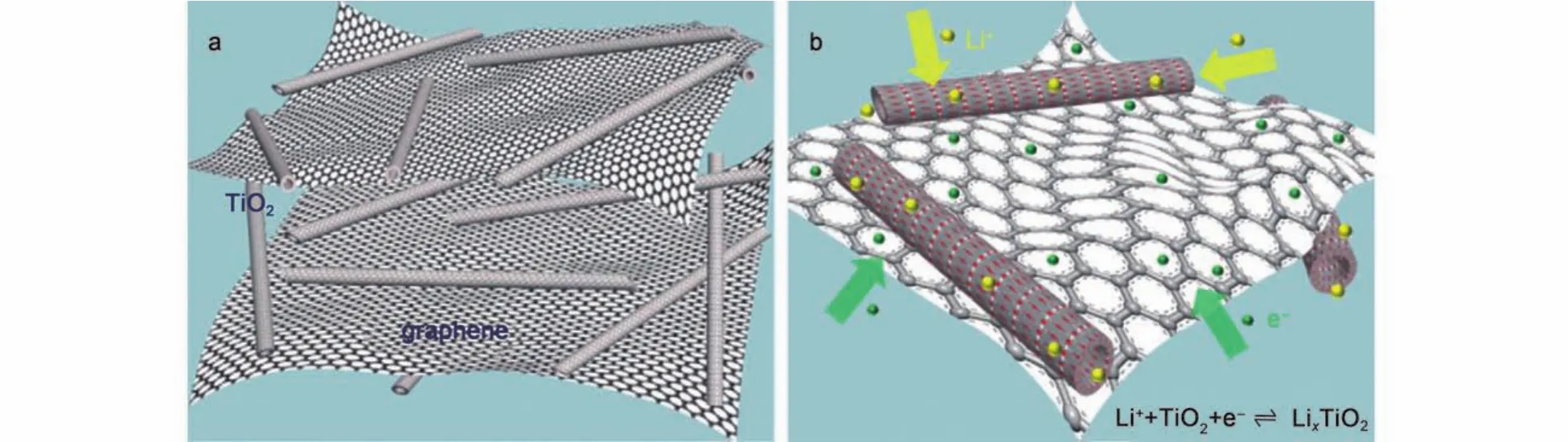
图6 TNTs与石墨烯复合示意图Fig.6 Illustrations of the TiO2nanotubes/graphene composite
Wang等128制备了TNT与石墨烯的复合材料(图6),这种复合物是一种比表面积很大的多孔材料,与电解液接触面积大,增强了锂离子的传导.石墨烯包裹TNT的同时,TNT在石墨烯层间的穿插防止了石墨烯的凝聚结块,使两种材料的性能均得到了提升.同时,在材料制备过程中,TNT和石墨烯是在水热反应中同时形成的,它们之间形成Ti-O-C化学键,石墨烯的π键系统充当了电子储存层,从而使电子可以快速传导,在大电流充放电循环中仍有良好的电化学性能:在4000 mA·g–1的电流下恒流充放电循环50圈后,比容量仍有150 mAh·g–1;在8000 mA·g–1的电流下恒流充放电循环2000圈后,比容量仍维持在80 mAh·g–1.在TNTs与石墨烯复合材料中,TNTs与石墨烯的连接方式对性能有很大的影响.Tang等129在水热反应前将TiO2的胶体涂覆在氧化石墨烯的表面,所得TNTs/石墨烯复合物中TNTs断面与石墨烯接触,其性能明显不及TNTs管壁与石墨烯接触的复合材料.
4.4.2 与金属氧化物的复合
尽管电导率不及碳纳米材料,但一些金属氧化物因其较高的理论比容量、高密度等优点,仍受到了不少研究人员的关注,成为制备TNTs复合材料的一种不错选择.Zhu等130制备了SnO2纳米片和TNTs的复合材料.SnO2比容量较高(782 mAh·g–1),但导电性差,脱嵌锂过程中体积变化大(300%),循环稳定性差.通过两者复合,材料兼具SnO2比容量大、TNTs循环性能好的特点,在1600 mA·g–1的电流密度下,循环30圈后,材料的比容量为530 mAh·g–1.
Ortiz等131利用TNTs阵列激活并在电解池中导向生长铁微晶,制得了Fe2O3纳米线和TNTs复合物,这种复合物具有较大的比表面积,且能有效避免大电流充放电时电极结构的塌陷.在1C倍率下充放电,面积比容量为240 µAh·cm–2,显示出良好的电化学性能.
Kim等132通过物理混合的方法制得了TNTs和钛酸锂(Li4Ti5O12,LTO)纳米颗粒的混合物,混合物中LTO充当的TNTs骨架,防止充放电循环中TNTs的塌陷或凝聚,提升了材料的比容量.通过调节TNTs和LTO的比例,发现当TNTs:LTO质量比为2:8时,材料密度(1.22 g·cm–3)达到最大,质量比容量为纯TNTs的三倍之多.
当然,这种方法本身也存在着一些不足.混合物分布不均,易破坏TNTs原有结构等问题给材料的电化学性能带来了负面的影响.如何有效地解决这些问题也成为研究人员下一步研究的课题.
5 结论与展望
TiO2作为锂离子电池负极材料,展现出良好的循环稳定性、安全性和环境友好性,但较低的比容量和导电性限制了它的实际应用.通过制备纳米结构的TNTs,材料的导电性和比容量得到了明显提升.形貌和晶型对TNTs电化学性能有直接影响,通过调整制备过程中的工艺参数(反应时间和温度、pH值、起始物质、电解液浓度和组分等),可以调控其微观形貌,进而调控其电化学性能.同时,结构设计、掺杂、负载其他材料、制备混合材料等方法也可明显提高材料导电性及比容量,进而提升其电化学性能.其中,涂覆碳膜的TNTs在7500 mA·g–1的电流密度下比容量为150 mAh·g–1,TNTs与SnO2的复合材料比容量大于500 mAh·g–1同时也具有良好的循环稳定性和倍率性能.
然而,掺杂、结构设计等方法尽管改善材料导电性,进而提升了循环稳定性,但难以大幅度提升材料比容量;负载其他材料、制备混合材料尽管可以在一定程度上提升材料比容量,但制备方法通常较为复杂,材料均一性差,难以适应大规模的工业生产.另一方面,和其他金属氧化物锂离子电池负极材料一样,TNTs也具有首圈库伦效率较低的问题,而目前的研究重点主要集中在提升TNTs比容量上,对提升其首圈库伦效率的研究较少涉及.相信随着研究不断深入展开,这些问题将在不久的将来得到解决,从而使TNTs锂离子电池负极材料得到广泛的实际应用.
(1)Scrosati,B.Nature 1995,373(6515),557.doi:10.1038/ 373557a0
(2)Tarascon,J.M.;Armand,M.Nature 2001,414(6861),359.doi:10.1038/35104644
(3)Bavykin,D.V.;Friedrich,J.M.;Walsh,F.C.Adv.Mater.2006,18(21),2807.
(4)Ortiz,G.F.;Hanzu,I.;Djenizian,T.;Lavela,P.;Tirado,J.L.;Knauth,P.Chem.Mater.2009,21(1),63.doi:10.1021/ cm801670u
(5)Djenizian,T.;Hanzu,I.;Knauth,P.J.Mater.Chem.2011,21(27),9925.doi:10.1039/c0jm04205f
(6)Arico,A.S.;Bruce,P.;Scrosati,B.;Tarascon,J.M.;Van Schalkwijk,W.Nat.Mater.2005,4(5),366.doi:10.1038/ nmat1368
(7)Armstrong,A.R.;Armstrong,G.;Canales,J.;Garcia,R.;Bruce,P.G.Adv.Mater.2005,17(7),862.
(8)Gentili,V.;Brutti,S.;Hardwick,L.J.;Armstrong,A.R.;Panero,S.;Bruce,P.G.Chem.Mater.2012,24(22),4468.doi:10.1021/cm302912f
(9)Kasuga,T.;Hiramatsu,M.;Hoson,A.;Sekino,T.;Niihara,K.Langmuir 1998,14(12),3160.doi:10.1021/la9713816
(10)Chen,Q.;Du,G.H.;Zhang,S.;Peng,L.M.Acta Crystallographica Section B-Structural Science 2002,58,587.doi:10.1107/S0108768102009084
(11)Huang,J.P.;Yuan,D.D.;Zhang,H.Z.;Cao,Y.L.;Li,G.R.;Yang,H.X.;Gao,X.P.RSC Advances 2013,3(31),12593.doi:10.1039/c3ra42413h
(12)Kim,H.S.;Yu,S.H.;Sung,Y.E.;Kang,S.H.J.Alloy.Compd.2014,597,275.doi:10.1016/j.jallcom.2014.02.013
(13)Xu,X.;Fan,Z.;Ding,S.;Yu,D.;Du,Y.Nanoscale 2014,6(10),5245.doi:10.1039/c3nr06736j
(14)Bavykin,D.V.;Parmon,V.N.;Lapkin,A.A.;Walsh,F.C.J.Mater.Chem.2004,14(22),3370.doi:10.1039/b406378c
(15)Kasuga,T.;Hiramatsu,M.;Hoson,A.;Sekino,T.;Niihara,K.Adv.Mater.1999,11(15),1307.
(16)Choi,M.G.;Lee,Y.G.;Song,S.W.;Kim,K.M.Electrochim.Acta 2010,55(20),5975.doi:10.1016/ j.electacta.2010.05.052
(17)Gajovic,A.;Friscic,I.;Plodinec,M.;Ivekovic,D.J.Mol.Struct.2009,924–926,183.
(18)Morgan,D.L.;Triani,G.;Blackford,M.G.;Raftery,N.A.;Frost,R.L.;Waclawik,E.R.J.Mater.Sci.2011,46(2),548.doi:10.1007/s10853-010-5016-0
(19)Zhang,Q.H.;Gao,L.;Zheng,S.;Sun,J.Acta Chim.Sin.2002,60(8),1439. [张青红,高 濂,郑 珊,孙 静.化学学报,2002,60(8),1439]
(20)Seo,H.K.;Kim,G.S.;Ansari,S.G.;Kim,Y.S.;Shin,H.S.;Shim,K.H.;Suh,E.K.Sol.Energy Mater.Sol.Cells 2008,92(11),1533.doi:10.1016/j.solmat.2008.06.019
(21)Suzuki,Y.;Pavasupree,S.;Yoshikawa,S.;Kawahata,R.J.Mater.Res.2005,20(4),1063.doi:10.1557/JMR.2005.0135
(22)Ma,R.Z.;Fukuda,K.;Sasaki,T.;Osada,M.;Bando,Y.J.Phys.Chem.B 2005,109(13),6210.doi:10.1021/ jp044282r
(23)Yuan,Z.Y.;Su,B.L.Colloids and Surfaces APhysicochemical and Engineering Aspects 2004,241(1–3),173.doi:10.1016/j.colsurfa.2004.04.030
(24)Sun,X.M.;Li,Y.D.Chemistry-A European Journal 2003,9(10),2229.doi:10.1002/chem.200204394
(25)Ma,R.Z.;Bando,Y.;Sasaki,T.Chem.Phys.Lett.2003,380(5–6),577.doi:10.1016/j.cplett.2003.09.069
(26)Tsai,C.C.;Nian,J.N.;Teng,H.S.Appl.Surf.Sci.2006,253(4),1898.doi:10.1016/j.apsusc.2006.03.035
(27)Du,G.H.;Chen,Q.;Che,R.C.;Yuan,Z.Y.;Peng,L.M.Appl.Phys.Lett.2001,79(22),3702.doi:10.1063/ 1.1423403
(28)Wang,W.Z.;Varghese,O.K.;Paulose,M.;Grimes,C.A.;Wang,Q.L.;Dickey,E.C.J.Mater.Res.2004,19(2),417.doi:10.1557/jmr.2004.19.2.417
(29)Yao,B.D.;Chan,Y.F.;Zhang,X.Y.;Zhang,W.F.;Yang,Z.Y.;Wang,N.Appl.Phys.Lett.2003,82(2),281.doi:10.1063/ 1.1537518
(30)Menzel,R.;Peiro,A.M.;Durrant,J.R.;Shaffer,M.S.P.Chem.Mater.2006,18(25),6059.doi:10.1021/cm061721l
(31)Morgado,E.;de Abreu,M.A.S.;Pravia,O.R.C.;Marinkovic,B.A.;Jardim,P.M.;Rizzo,F.C.;Araujo,A.S.Solid State Sci.2006,8(8),888.doi:10.1016/ j.solidstatesciences.2006.02.039
(32)Kukovecz,A.;Hodos,N.;Horvath,E.;Radnoczi,G.;Konya,Z.;Kiricsi,I.J.Phys.Chem.B 2005,109(38),17781.doi:10.1021/jp054320m
(33)Nosheen,S.;Galasso,F.S.;Suib,S.L.Langmuir 2009,25(13),7623.doi:10.1021/la9002719
(34)Yang,J.J.;Jin,Z.S.;Wang,X.D.;Li,W.;Zhang,J.W.;Zhang,S.L.;Guo,X.Y.;Zhang,Z.J.Dalton Trans.2003,No.20,3898.
(35)Gao,T.;Fjellvag,H.;Norby,P.Inorg.Chem.2009,48(4),1423.doi:10.1021/ic801508k
(36)Liu,N.;Chen,X.;Zhang,J.;Schwank,J.W.Catal.Today 2014,225(0),34.
(37)Lai,Y.K.;Sun,L.;Zun,J.;Lin,C.J.Acta Phys.-Chim.Sin.2004,20,1063. [赖跃坤,孙 岚,左 娟,林昌健.物理化学学报,2004,20,1063.] doi:10.3866/ PKU.WHXB 20040901
(38)Macak,J.M.;Tsuchiya,H.;Taveira,L.;Aldabergerova,S.;Schmuki,P.Angew.Chem.Int.Edit.2005,44(45),7463.
(39)Pervez,S.;Kim,D.;Doh,C.H.;Farooq,U.;Yaqub,A.;Choi,J.H.;Lee,Y.J.;Saleem,M.Mater.Lett.2014,137,347.doi:10.1016/j.matlet.2014.09.032
(40)John,S.E.;Mohapatra,S.K.;Misra,M.Langmuir 2009,25(14),8240.doi:10.1021/la900426j
(41)Li,H.;Martha,S.K.;Unocic,R.R.;Luo,H.;Dai,S.;Qu,J.J.Power Sources 2012,218,88.doi:10.1016/j.jpowsour.2012.06.096
(42)Lakshmi,B.B.;Dorhout,P.K.;Martin,C.R.Chem.Mater.1997,9(3),857.doi:10.1021/cm9605577
(43)Sudant,G.;Baudrin,E.;Larcher,D.;Tarascon,J.M.J.Mater.Chem.2005,15(12),1263.
(44)Kavan,L.;Grätzel,M.;Gilbert,S.E.;Klemenz,C.;Scheel,H.J.J.Am.Chem.Soc.1996,118(28),6716.doi:10.1021/ ja954172l
(45)Exnar,I.;Kavan,L.;Huang,S.Y.;Grätzel,M.J.Power Sources 1997,68(2),720.doi:10.1016/S0378-7753(96) 02581-5
(46)Lindstrom,H.;Sodergren,S.;Solbrand,A.;Rensmo,H.;Hjelm,J.;Hagfeldt,A.;Lindquist,S.E.J.Phys.Chem.B 1997,101(39),7717.doi:10.1021/jp970490q
(47)Borghols,W.J.H.;Lutzenkirchen-Hecht,D.;Haake,U.;van Eck,E.R.H.;Mulder,F.M.;Wagemaker,M.Phys.Chem.Chem.Phys.2009,11(27),5742.doi:10.1039/b823142g
(48)Lafont,U.;Carta,D.;Mountjoy,G.;Chadwick,A.V.;Kelder,E.M.J.Phys.Chem.C 2010,114(2),1372.doi:10.1021/ jp908786t
(49)Wagemaker,M.;Kentgens,A.P.M.;Mulder,F.M.Nature 2002,418(6896),397.doi:10.1038/nature00901
(50)Wagemaker,M.;Borghols,W.J.H.;van Eck,E.R.H.;Kentgens,A.P.M.;Kearley,G.L.;Mulder,F.M.Chemistry-A European Journal 2007,13(7),2023.
(51)Macklin,W.J.;Neat,R.J.Solid State Ionics 1992,53,694.
(52)Hu,Y.S.;Kienle,L.;Guo,Y.G.;Maier,J.Adv.Mater.2006,18(11),1421.
(53)Reddy,M.A.;Kishore,M.S.;Pralong,V.;Caignaert,V.;Varadaraju,U.V.;Raveau,B.Electrochem.Commun.2006,8(8),1299.doi:10.1016/j.elecom.2006.05.021
(54)Marinaro,M.;Pfanzelt,M.;Kubiak,P.;Marassi,R.;Wohlfahrt-Mehrens,M.J.Power Sources 2011,196(22),9825.doi:10.1016/j.jpowsour.2011.07.008
(55)Zukalova,M.;Kalbac,M.;Kavan,L.;Exnar,I.;Gräetzel,M.Chem.Mater.2005,17(5),1248.doi:10.1021/cm048249t
(56)Ryu,W.H.;Nam,D.H.;Ko,Y.S.;Kim,R.H.;Kwon,H.S.Electrochim.Acta 2012,61,19.doi:10.1016/j.electacta.2011.11.042
(57)Fang,H.T.;Liu,M.;Wang,D.W.;Sun,T.;Guan,D.S.;Li,F.;Zhou,J.G.;Sham,T.K.;Cheng,H.M.Nanotechnology 2009,20(22).266.
(58)Kim,J.;Cho,J.J.Electrochem.Soc.2007,154(6),A542.
(59)Koudriachova,M.V.;Harrison,N.M.;de Leeuw,S.W.Phys.Rev.Lett.2001,86(7),1275.doi:10.1103/ PhysRevLett.86.1275
(60)Yang,H.G.;Sun,C.H.;Qiao,S.Z.;Zou,J.;Liu,G.;Smith,S.C.;Cheng,H.M.;Lu,G.Q.Nature 2008,453(7195),638.doi:10.1038/nature06964
(61)Chen,J.S.;Tan,Y.L.;Li,C.M.;Cheah,Y.L.;Luan,D.Y.;Madhavi,S.;Boey,F.Y.C.;Archer,L.A.;Lou,X.W.J.Am.Chem.Soc.2010,132(17),6124.doi:10.1021/ ja100102y
(62)Pan,D.;Huang,H.;Wang,X.;Wang,L.;Liao,H.;Li,Z.;Wu,M.Journal of Materials Chemistry A 2014,2(29),11454.doi:http://dx.doi.org/10.1039/c4ta01613k
(63)Yan,J.Y.;Song,H.H.;Yang,S.B.;Chen,X.H.Mater.Chem.Phys.2009,118(2–3),367.doi:10.1016/ j.matchemphys.2009.08.007
(64)Albu,S.P.;Ghicov,A.;Aldabergenova,S.;Drechsel,P.;LeClere,D.;Thompson,G.E.;Macak,J.M.;Schmuki,P.Adv.Mater.2008,20(21),4135.
(65)Lamberti,A.;Garino,N.;Sacco,A.;Bianco,S.;Manfredi,D.;Gerbaldi,C.Electrochim.Acta 2013,102(0),233.
(66)Lamberti,A.;Garino,N.;Sacco,A.;Bianco,S.;Chiodoni,A.;Gerbaldi,C.Electrochim.Acta 2015,151,222.doi:10.1016/j.electacta.2014.10.150
(67)Guan,D.S.;Cai,C.A.;Wang,Y.J.Nanosci.Nanotechnol.2011,11(4),3641.doi:10.1166/jnn.2011.3765
(68)Panda,S.K.;Yoon,Y.;Jung,H.S.;Yoon,W.S.;Shin,H.J.Power Sources 2012,204,162.doi:10.1016/j.jpowsour.2011.12.048
(69)Gonzalez,J.R.;Alcantara,R.;Nacimiento,F.;Ortiz,G.F.;Tirado,J.L.;Zhecheva,E.;Stoyanova,R.J.Phys.Chem.C 2012,116(38),20182.doi:10.1021/jp3050115
(70)Freitas,R.G.;Justo,S.G.;Pereira,E.C.J.Power Sources 2013,243,569.doi:10.1016/j.jpowsour.2013.06.044
(71)Han,H.;Song,T.;Lee,E.K.;Devadoss,A.;Jeon,Y.;Ha,J.;Chung,Y.C.;Choi,Y.M.;Jung,Y.G.;Paik,U.ACS Nano 2012,6(9),8308.doi:10.1021/nn303002u
(72)Beguin,F.;Chevallier,F.;Vix,C.;Saadallah,S.;Rouzaud,J.N.;Frackowiak,E.J.Phys.Chem.Solids 2004,65(2–3),211.doi:10.1016/j.jpcs.2003.10.050
(73)Li,H.;Shi,L.H.;Lu,W.;Huang,X.J.;Chen,L.Q.J.Electrochem.Soc.2001,148(8),A915.
(74)Aurbach,D.;Weissman,I.;Zaban,A.;Dan,P.Electrochim.Acta 1999,45(7),1135.doi:10.1016/S0013-4686(99)00312-6
(75)Aurbach,D.Electrochim.Acta 1999,45(1–2),1.doi:10.1016/S0013-4686(99)00188-7
(76)Verma,P.;Maire,P.;Novak,P.Electrochim.Acta 2010,55(22),6332.doi:10.1016/j.electacta.2010.05.072
(77)Kawamura,T.;Okada,S.;Yamaki,J.J.Power Sources 2006,156(2),547.doi:10.1016/j.jpowsour.2005.05.084
(78)Brutti,S.;Gentili,V.;Menard,H.;Scrosati,B.;Bruce,P.G.Adv.Energy Mater.2012,2(3),322.doi:10.1002/aenm.201100492
(79)Morterra,C.Journal of the Chemical Society-Faraday Transactions I 1988,84,1617.doi:10.1039/f19888401617
(80)Li,C.;Zhang,H.P.;Fu,L.J.;Liu,H.;Wu,Y.P.;Ram,E.;Holze,R.;Wu,H.Q.Electrochim.Acta 2006,51(19),3872.doi:10.1016/j.electacta.2005.11.015
(81)Kim,M.G.;Kim,H.;Cho,J.J.Electrochem.Soc.2010,157(7),A802.
(82)Bi,Z.;Paranthaman,M.P.;Menchhofer,P.A.;Dehoff,R.R.;Bridges,C.A.;Chi,M.;Guo,B.;Sun,X.G.;Dai,S.J.Power Sources 2013,222,461.doi:10.1016/j.jpowsour.2012.09.019
(83)Zhang,Z.J.;Zeng,Q.Y.;Chou,S.L.;Li,X.J.;Li,H.J.;Ozawa,K.;Liu,H.K.;Wang,J.Z.Electrochim.Acta 2014,133,570.doi:10.1016/j.electacta.2014.04.049
(84)Zhao,B.;Jiang,S.;Su,C.;Cai,R.;Ran,R.;Tade,M.O.;Shao,Z.Journal of Materials Chemistry A 2013,1(39),12310.doi:10.1039/c3ta12770b
(85)Chen,J.;Yang,L.;Tang,Y.J.Power Sources 2010,195(19),6893.doi:10.1016/j.jpowsour.2010.04.005
(86)Song,T.;Han,H.;Choi,H.;Lee,J.W.;Park,H.;Lee,S.;Park,W.I.;Kim,S.;Liu,L.;Paik,U.Nano Research 2014,7(4),491.doi:10.1007/s12274-014-0415-1
(87)Lu,X.;Wang,G.;Zhai,T.;Yu,M.;Gan,J.;Tong,Y.;Li,Y.Nano Lett.2012,12(3),1690.doi:10.1021/nl300173j
(88)Wu,H.;Xu,C.;Xu,J.;Lu,L.;Fan,Z.;Chen,X.;Song,Y.;Li,D.Nanotechnology 2013,24(45).
(89)Salari,M.;Konstantinov,K.;Liu,H.K.J.Mater.Chem.2011,21(13),5128.doi:10.1039/c0jm04085a
(90)Guo,W.;Xue,X.;Wang,S.;Lin,C.;Wang,Z.L.Nano Lett.2012,12(5),2520.doi:10.1021/nl3007159
(91)Li,Z.;Ding,Y.;Kang,W.;Li,C.;Lin,D.;Wang,X.;Chen,Z.;Wu,M.;Pan,D.Electrochim.Acta 2015,161,40.
(92)Hoang,S.;Berglund,S.P.;Hahn,N.T.;Bard,A.J.;Mullins,C.B.J.Am.Chem.Soc.2012,134(8),3659.doi:10.1021/ ja211369s
(93)Zhang,J.;Zhang,J.;Ren,H.;Yu,L.;Wu,Z.;Zhang,Z.J.Alloy.Compd.2014,609,178.doi:10.1016/j.jallcom.2014.04.115
(94)Li,Y.;Wang,Z.;Lv,X.J.Journal of Materials Chemistry A 2014,2(37),15473.doi:10.1039/C4TA02890B
(95)Xu,J.;Wang,Y.;Li,Z.;Zhang,W.J.Power Sources 2008,175(2),903.doi:10.1016/j.jpowsour.2007.10.014
(96)Kyeremateng,N.A.;Vacandio,F.;Sougrati,M.T.;Martinez,H.;Jumas,J.C.;Knauth,P.;Djenizian,T.J.Power Sources 2013,224,269.doi:10.1016/j.jpowsour.2012.09.104
(97)Pfanzelt,M.;Kubiak,P.;Fleischhammer,M.;Wohlfahrt-Mehrens,M.J.Power Sources 2011,196(16),6815.doi:10.1016/j.jpowsour.2010.09.109
(98)Fan,J.;Zhao,Z.;Wang,J.;Zhu,L.Appl.Surf.Sci.2015,324(0),691.
(99)Cheng,Z.W.;Feng,L.;Chen,J.M.;Yu,J.M.;Jiang,Y.F.J.Hazard.Mater.2013,254,354.
(100)Liu,H.J.;Liu,G.G.;Zhou,Q.X.;Xie,G.H.;Hou,Z.H.;Zhang,M.L.;He,Z.W.Microporous Mesoporous Mat.2011,142(2–3),439.doi:10.1016/j.micromeso.2010.11.035
(101)Fan,X.;Wan,J.;Liu,E.;Sun,L.;Hu,Y.;Li,H.;Hu,X.;Fan,J.Ceram.Int.2015,41(3,Part B),5107.doi:10.1016/j.ceramint.2014.12.083
(102)Fan,X.;Fan,J.;Hu,X.;Liu,E.;Kang,L.;Tang,C.;Ma,Y.;Wu,H.;Li,Y.Ceram.Int.2014,40(10),15907.doi:10.1016/j.ceramint.2014.07.119
(103)Li,Y.;Wang,Y.;Kong,J.;Wang,J.Appl.Surf.Sci.2015,328(0),115.
(104)Han,W.Q.;Wen,W.;Yi,D.;Liu,Z.;Maye,M.M.;Lewis,L.;Hanson,J.;Gang,O.J.Phys.Chem.C 2007,111(39),14339.doi:10.1021/jp074381f
(105)Yu,Y.;Wu,H.H.;Zhu,B.L.;Wang,S.R.;Huang,W.P.;Wu,S.H.;Zhang,S.M.Catal.Lett.2008,121(1–2),165.doi:10.1007/s10562-007-9316-1
(106)Umek,P.;Pregelj,M.;Gloter,A.;Cevc,P.;Jaglicic,Z.;Ceh,M.;Pirnat,U.;Arcon,D.J.Phys.Chem.C 2008,112(39),15311.doi:10.1021/jp805005k
(107)Meksi,M.;Berhault,G.;Guillard,C.;Kochkar,H.Catal.Commun.2015,61,107.doi:10.1016/j.catcom.2014.12.020
(108)Kim,D.H.;Jang,J.S.;Goo,N.H.;Kwon,M.S.;Lee,J.W.;Choi,S.H.;Shin,D.W.;Kim,S.J.;Lee,K.S.Catal.Today 2009,146(1–2),230.doi:10.1016/j.cattod.2009.04.007
(109)Szirmai,P.;Horvath,E.;Nafradi,B.;Mickovic,Z.;Smajda,R.;Djokic,D.M.;Schenk,K.;Forro,L.;Magrez,A.J.Phys.Chem.C 2013,117(1),697.doi:10.1021/jp3104722
(110)Long,L.Z.;Wu,L.P.;Yang,X.;Li,X.J.Journal of Materials Science &Technology 2014,30(8),765.doi:10.1016/j.jmst.2014.03.010
(111)Kim,H.S.;Kang,S.H.;Chung,Y.H.;Sung,Y.E.Electrochem.Solid State Lett.2010,13(2),A15.
(112)Ivanov,S.;Grieseler,R.;Cheng,L.;Schaaf,P.;Bund,A.J.Electroanal.Chem.2014,731,6.doi:10.1016/j.jelechem.2014.07.038
(113)Brumbarov,J.;Kunze-Liebhaeuser,J.J.Power Sources 2014,258,129.doi:10.1016/j.jpowsour.2014.02.049
(114)Guan,D.;Li,J.;Gao,X.;Yuan,C.J.Power Sources 2014,246,305.doi:10.1016/j.jpowsour.2013.07.096
(115)Fan,Y.;Zhang,N.;Zhang,L.;Shao,H.;Wang,J.;Zhang,J.;Cao,C.Electrochim.Acta 2013,94,285.doi:10.1016/ j.electacta.2013.01.114
(116)Kulova,T.L.;Skundin,A.M.;Pleskov,Y.V.;Terukov,E.I.;Kon'kov,O.I.J.Electroanal.Chem.2007,600(1),217.doi:10.1016/j.jelechem.2006.07.002
(117)Wu,X.M.;Zhang,S.C.;Wang,L.L.;Du,Z.J.;Fang,H.;Ling,Y.H.;Huang,Z.H.J.Mater.Chem.2012,22(22),11151.doi:10.1039/c2jm30885a
(118)Jeun,J.H.;Park,K.Y.;Kim,D.H.;Kim,W.S.;Kim,H.C.;Lee,B.S.;Kim,H.;Yu,W.R.;Kang,K.;Hong,S.H.Nanoscale 2013,5(18),8480.doi:10.1039/c3nr01964k
(119)Fan,Y.;Zhang,N.;Zhang,L.;Shao,H.;Wang,J.;Zhang,J.;Cao,C.Electrochim.Acta 2013,94(0),285.
(120)Kang,K.Y.;Lee,Y.G.;Kim,S.;Seo,S.R.;Kim,J.C.;Kim,K.M.Mater.Chem.Phys.2012,137(1),169.doi:10.1016/j.matchemphys.2012.09.001
(121)Park,S.J.;Kim,Y.J.;Lee,H.J.Power Sources 2011,196(11),5133.doi:10.1016/j.jpowsour.2011.01.105
(122)Bresser,D.;Oschmann,B.;Tahir,M.N.;Mueller,F.;Lieberwirth,I.;Tremel,W.;Zentel,R.;Passerini,S.J.Electrochem.Soc.2015,162(2),A3013.
(123)Plylahan,N.;Letiche,M.;Samy Barr,M.K.;Ellis,B.;Maria,S.;Phan,T.N.T.;Bloch,E.;Knauth,P.;Djenizian,T.J.Power Sources 2015,273,1182.
(124)He,B.L.;Dong,B.;Li,H.L.Electrochem.Commun.2007,9(3),425.doi:10.1016/j.elecom.2006.10.008
(125)Fang,D.;Huang,K.L.;Liu,S.Q.;Li,Z.J.J.Alloy.Compd.2008,464(1–2),L5.
(126)Kim,H.;Kim,M.G.;Shin,T.J.;Shin,H.J.;Cho,J.Electrochem.Commun.2008,10(11),1669.doi:10.1016/ j.elecom.2008.08.035
(127)Kang,K.Y.;Shin,D.O.;Lee,Y.G.;Kim,S.;Kim,K.M.Journal of Electroceramics 2014,32(2–3),246.doi:10.1007/s10832-013-9882-0
(128)Wang,J.;Zhou,Y.K.;Xiong,B.;Zhao,Y.Y.;Huang,X.J.;Shao,Z.P.Electrochim.Acta 2013,88,847.doi:10.1016/ j.electacta.2012.10.010
(129)Tang,Y.;Liu,Z.;Lu,X.;Wang,B.;Huang,F.RSC Advances 2014,4(68),36372.doi:10.1039/C4RA05027D
(130)Zhu,C.;Xia,X.;Liu,J.;Fan,Z.;Chao,D.;Zhang,H.;Fan,H.J.Nano Energy 2014,4,105.
(131)Ortiz,G.F.;Hanzu,I.;Lavela,P.;Tirado,J.L.;Knauth,P.;Djenizian,T.J.Mater.Chem.2010,20(20),4041.doi:10.1039/b927122h
(132)Kim,K.M.;Kang,K.Y.;Kim,S.;Lee,Y.G.Current Applied Physics 2012,12(4),1199.doi:10.1016/j.cap.2012.02.059
TiO2Nanotubes as an Anode Material for Lithium Ion Batteries
WANG Qian-Wen1DU Xian-Feng1,2,*CHEN Xi-Zi1XU You-Long1,2
(1Electronic Materials Research Laboratory,Key Laboratory of the Ministry of Education,Xi'an Jiaotong University,Xi'an 710049,P.R.China;2International Center of Dielectric Research,Xi'an Jiaotong University,Xi'an 710049,P.R.China)
In recent years,TiO2has been widely investigated as a promising anode material for lithium ion batteries because of its low volume change during the charge/discharge process,environmental benignity,and high safet·y.However,it suffers from poor electron transport,slow ion diffusion,and low theoretical capacity(335 mAhg–1),which limit its practical application.In this paper,we review the development history and latest progress of TiO2nanotubes(TNTs) as anode materials.Three typical synthesis methods of TNTs,namely,hydrothermal method,anodic oxidation,and template method,are analyzed in detail.We explain the formation mechanism,compare the advantages and disadvantages of each method,and identify the factors influencing the formation of TNTs.We also carefully analyze the morphology and crystallography of TNTs and describe how they influence the electrochemical performance.It is pointed out that c-axis oriented,arrayed,unsealed TNTs with a wall thickness less than 5 nm show better electrochemical performance.Various approaches for improving the electrochemical performance of TNTs are summarized,including preparation of threedimensional(3D) structured electrodes,doping,coating,and synthesis of composites.Among these approaches,compositing with materials that have high capacity and high conductivity has proven to be effective,convenient,and controllable.The achievements and the problems associated with each approach are summarized,and the possible research directions and prospects of TNTs as anode materials for Li-ion batteries in the future are discussed.
April 20,2015;Revised:June 15,2015;Published on Web:June 16,2015.
O646
iew]
10.3866/PKU.WHXB201506162 www.whxb.pku.edu.cn
*Corresponding author.Email:xianfengdu@mail.xjtu.edu.cn;Tel:+86-29-82668669-82.
The project was supported by the Natural Science Foundation of Shaanxi Province,China(2014JM6231),Scientific Research Foundation for the Returned Overseas Chinese Scholars,State Education Ministry,and Fundamental Research Funds for the Central Universities,China(XJJ2012076).陕西省自然科学基金项目(2014JM6231),教育部留学回国人员启动基金项目及中央高校基本科研业务费专项资金(XJJ2012076)资助© Editorial office of Acta Physico-Chimica Sinica
