固载型杂化微球模拟催化氧化脱硫性能
2015-08-19宋少飞周福林弓巧娟沈淑坤
宋少飞,周福林,弓巧娟,沈淑坤
(1运城学院应用化学系,山西 运城 044000;2陕西师范大学材料科学与工程学院,陕西 西安 710062)
近年来,环境污染的问题日趋严重,已经引起了世界各国的普遍关注。相关研究表明,机动车尾气排放是造成空气质量下降的主要因素。为了提高油品的质量,从根本上降低尾气排放带来的危害,国际上陆续制定了严格的含硫尾气排放标准[1-2]。与此相应,燃料油脱硫技术也应运而生,如加氢脱硫已实现工业化应用。但加氢脱硫技术对大分子的杂环含硫化合物(如二苯并噻吩及其衍生物)无法实现有效脱除,不能达到深度脱硫的目的。因此,燃油深度脱硫技术研究已成为石油加工领域非常活跃的研究课题。
当前,吸附法[3-4]、氧化法[5-6]以及生物化学法[7-8]等非加氢脱硫技术得到广泛研究,以期实现工业化程度的深度脱硫。其中,以绿色氧化剂过氧化氢为氧源、杂多酸及其盐为催化剂的催化氧化脱硫体系被广泛认为是一种最具优势的超深度催化脱硫技术。燃油中的硫化物(噻吩、二苯并噻吩及其衍生物等)在该体系中被选择性地氧化为砜,然后利用液-液萃取手段将极性较强的砜除去,具有操作简便、催化活性高、无二次污染等优势。如Lucy等[9]设计了一种磷钨酸-过氧化氢体系,对二苯并噻吩进行了催化氧化研究。结果表明,最佳条件下目标分子转化率达到了100%。将此项研究应用到汽油脱硫工艺中,可以使硫化物全部氧化。但研究发现,过氧化氢的分解与二苯并噻吩的转化之间存在竞争,导致该工艺在实现工业化应用方面还略有不足:①氧化剂使用效率较低;②催化剂回收再利用困难;③硫化物氧化选择性较差;④易引入其他成分。我国科学家在深度催化氧化脱硫方面也做了大量工作,如Meng等[10]成功合成了一系列磷酸咪唑盐型离子液体,以其为溶剂,使用七钼酸铵四水合物为催化剂,过氧化氢为氧化剂,对4,6-二甲基二苯并噻吩进行了催化氧化。结果表明,该体系表现出了非常高的催化性能,在50℃反应3h,4,6-二甲基二苯并噻吩的转化率即可达到89.2%。另外,该体系反应条件温和,不影响燃料油品质,体系可以重复利用6次以上。
上述研究体系可以有效达到深度脱硫的要求,但在实现工业应用方面仍存在一定的限制,主要问题是反应后催化剂很难从体系中分离,一方面增加了催化剂的成本;另一方面造成了二次污染。而通过使用担载型催化剂,可有效解决这一难题,在已报道的研究工作中,担载型催化剂在催化氧化反应中已显示出明显的优势[11-12]。本文作者在正庚烷溶剂中,以PNIPAM高聚物微凝胶为模板,通过外源沉积技术制备了表面负载过氧磷钨杂多酸十六烷基季胺盐的杂化微球。利用SEM、FT-IR和TGA等技术对所得杂化微球进行了组成与形貌表征。然后,以合成的杂化微球为催化剂,30% H2O2为氧化剂,在十氢化萘、四氢化萘和正戊烷混合溶剂中,对硫化物噻吩、DBT以及4,6-二甲基二苯并噻吩进行了模拟催化氧化研究。结果表明,杂化微球对噻吩、DBT以及4,6-二甲基二苯并噻吩均具有较高的催化性能。其中,在最佳条件下,小分子噻吩可短时间内被完全氧化,DBT最大转化率达到了90%以上。而且,催化剂经处理后可循环利用3次以上。
1 实验部分
1.1 实验试剂
正庚烷,Span-80,过硫酸胺(APS),N,N,N',N'-四甲基乙二胺(TMED),N-异丙基丙烯酰胺(NIPAM),N,N'-亚甲基双丙稀酰胺(BA),丙酮,磷钨酸(HPW),十六烷基三甲基溴化胺(CTAB),30%过氧化氢,噻吩,二苯并噻酚(DBT),4,6-二甲基二苯并噻吩(4,6-DMDBT),乙腈,十氢化萘,四氢化萘和正戊烷,以上试剂均为分析纯。实验所用水均为去离子水。
1.2 实验方法
(1)PNIPAM微凝胶的制备 如已报道文献所述[13],将0.6g Span-80 加入到100mL正庚烷溶剂中,通入氮气进行保护,保持搅拌速度500r/min,20℃下充分乳化30min。称取1.2g NIPAM,1mL BA和0.5mL APS相继溶于6mL去离子水中,配成水相。将配成的水相加入到乳化液中,保持转速不变,继续搅拌10min,然后再移入1mL TMED,持续反应约4~5h。停止反应,固体产物用水和丙酮交替洗涤3~4次后,自然晾干。
(2)PW-HPW/PNIPAM担载型催化剂的制 备 量取80mL 正庚烷于150mL 的三口烧瓶中,然后加入1.5g 表面活性剂Span-80,通入氮气,将反应装置置于冰水浴中,搅拌至Span-80均匀分散。接着,将0.12g用HPW的过氧化氢溶液充分溶胀过夜的PNIPAM水凝胶移入瓶中。保持转速500r/min不变,再次搅拌30min后,用恒压漏斗以1滴/秒的速度加入CTAB的过氧化氢溶液。滴加完毕,转速不变,升高反应温度至50℃,持续反应4~5h。停止反应后,固体产物经二次水、丙酮交替洗涤3~4次后,室温下自然晾干。在相同条件下,不使用PNIPAM为模板,制备PW-HPW催化剂粉末,干燥后备用。
(3)模拟催化脱硫实验 分别量取20mL十氢萘、15mL四氢萘和15mL正戊烷,依次加入100mL的三口烧瓶中。然后,加入0.702mmol DBT(或同物质的量的噻吩或4,6-DMDBT)于瓶中,500r/min搅拌至完全溶解,再加入0.1g用30% H2O2溶胀的催化剂PW-HPW/PNIPAM。一定温度下,调整转速至1000r/min,持续反应4~5h。停止反应,过滤,使用丙酮和水将催化剂交替洗涤3~4次,室温下自然晾干备用。滤液取样进行GC-FID检测。另外,滤液用水与乙腈的混合溶剂萃取,即可将氧化产物砜除去。
1.3 表征
模板及杂化微球的形貌在PhilipXL-20型扫描电子显微镜上观察,并利用在线电子能谱进行元素分析,加速电压15kV或20kV。红外光谱在AVTAR360 Nicolet FTIR 光谱仪上测定。使用Perkin-Elmer TGA-7型热重分析仪对材料热稳定性及表面担载物含量进行测定。使用毛细管柱Agilen 6890N GC-FID气相色谱仪确定DBT的转化率。
2 结果与讨论
2.1 催化剂表征
对合成的微凝胶及杂化微球的形貌进行了扫描电镜观察,如图1所示。对于PNIPAM微凝胶[图1(a) 、(b)],可以看出,微凝胶呈规则的球形形貌,颗粒直径约70~90μm,表面光滑致密。在实验中发现,该微凝胶颗粒具有可逆的溶胀收缩过程。与PNIPAM模板相比,本实验所合成的表面沉积有杂多酸季铵盐的杂化微球也具有球形形貌[图1(c) 、(d)],单分散性较好,颗粒直径大约为150~200μm。不同的是,复合微球表面具有由小褶皱构成的规则形貌。这种微凝胶表面图案化的成因,已被大量实验所证实,来自于由模板的“限域沉积”现象,在胡道道小组[14-16]的相关报道中已被指出。
使用红外光谱对实验产物的组成进行了表征,结果如图2所示。在PNIPAM微凝胶红外谱图中,很明显,1665cm-1处的强吸收峰为C=O伸缩振动峰,胺基的伸缩振动峰位于3500~3300cm-1处[17-18]。与PNIPAM相比,PW-HPW/PNIPAM杂化微球的红外谱图中出现了明显的过氧磷钨酸季胺 盐的特征峰,875cm-1左右峰是(O—O)的振动峰,969cm-1出现的峰是(W=O)的振动峰[19]。根据文献报道,在540cm-1和604cm-1左右存在(O—W—O)的振动吸收峰[20],但本研究中,由于载体PNIPAM在该区域存在较强的吸收带,导致这两个峰表现得并不十分明显。

图1 PNIPAM微凝胶和PW-HPW/PNIPAM杂化微球的扫描电镜照片

图2 PW-HPW粉末、PNIPAM微凝胶和PW-HPW/PNIPAM杂化微球的红外光谱图
与红外表征结果一致,杂化微球表面的能谱分析也确证了杂多酸季铵盐被成功沉积于PNIPAM高聚物微凝胶表面,如图3所示。图中元素Au来自电镜观察前样品表面所镀的金膜;元素C、N和O主要为载体PNIPAM的组成元素,另外一部分C、N和O来自胺盐中的CTAB;而元素P和W则是PW-HPW特有的元素。此外,考虑到催化剂的热稳定性对其在工业应用方面具有非常重要的影响,使用热重分析仪对杂化微球的热分解行为进行了研究,如图4所示。杂化微球受热失重行为主要分为两个阶段,200℃前主要失去的是吸附水和溶剂,杂化微球从310℃开始受热分解,直至600℃分解完成,有机物被完全除去,残留物为WO3和P2O5。上述热分解行为表明杂化微球具有较高的热稳定性,可以承受300℃以下高温。
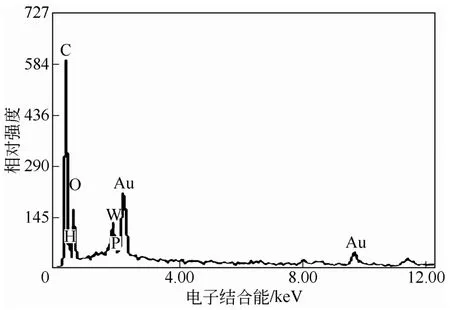
图3 表面能谱分析

图4 热重分析曲线
2.2 催化剂催化性能
2.2.1 反应温度和时间的影响
以合成的担载型杂化微球为催化剂,30% H2O2为氧化剂,其他条件与1.2节相同,分别在20℃、30℃、40℃和60℃下进行DBT催化氧化实验,通过不同时间取样测定DBT转化率,考察了温度和时间对催化剂催化效率的影响,如图5所示。可以看出,40℃之前,随着温度的升高,DBT转化率也随之升高。但随着进一步升高温度,60℃时催化剂效率略有降低。根据已有相关报道结果,升高温度在提高了催化剂催化效率的同时,也加速了氧化剂H2O2的分解,这应该是造成高温下DBT转化率下降的主要原因[21]。图5同时给出了反应时间对DBT催化氧化率的影响。曲线变化趋势表明反应初期DBT转化较快,反应后期,随着氧化剂被逐渐消耗,转化速度下降,反应8h后,催化反应基本完成,40℃时DBT最大转化率可达90%以上。
2.2.2 氧化剂用量的影响

图5 分别在20℃、30℃、40℃和60℃下DBT的转化率曲线
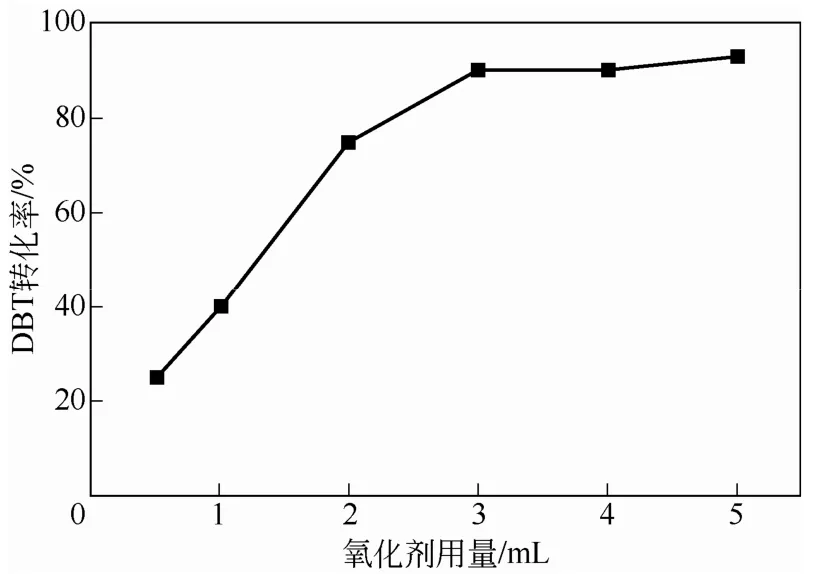
图6 氧化剂用量对DBT转化率的影响
通过使用不同体积的30% H2O2溶胀PW-HPW/PNIPAM杂化微球,在其他实验条件与1.2 节相同的情况下,对DBT进行了催化氧化实验,考察了氧化剂用量对DBT转化率的影响,如图6 所示。图中曲线变化表明,随氧化剂用量的增加,DBT转化率明显增加。过氧化氢用量为3mL时,转化率为90.32%。继续增加氧化剂用量,转化率略有增加,但增幅不大。实验中发现,杂化微球对过氧化氢的吸附存在极限,当氧化剂用量为3mL时,吸附饱和,此时增加氧化剂用量,过多的氧化剂将分散于反应体系,无法被杂化微球吸附。因此,氧化剂的用量以3mL为佳。
2.2.3 与非担载催化剂催化效率的对比
为了考察担载行为对催化剂催化性能的影响,保持其他反应条件不变,在40℃下,使用30% H2O2为氧化剂,DBT为硫化物,分别进行了无催化剂的空白实验和过氧磷钨杂多酸季胺盐粉末为催化剂的对比实验。同时,分别考察了杂化微球对硫化物噻吩和4,6-DMDBT的催化氧化性能,如图7所示。从图7中曲线变化趋势可以看出,空白试验条件下,DBT转化率几乎为零;非担载型催化剂催化效率最高,反应8h DBT几乎完全转化,表明固载后的杂化微球催化效率略低。但需要指出的是,非担载型催化剂反应后很难从体系回收,而杂化微球可以通 过离心从体系中分离,经处理后重复使用。另外,在对噻吩和4,6-DMDBT的催化氧化实验中,4,6-DMDBT转化能力略低于DBT,而噻吩6h后即可达到完全转化,表明小分子硫化物更易被催化氧化,与相关研究报道结果一致[22]。上述实验结果表明,表面担载过氧杂多酸的杂化微球对噻吩、DBT及4,6-DMDBT均具有较强的催化性能。
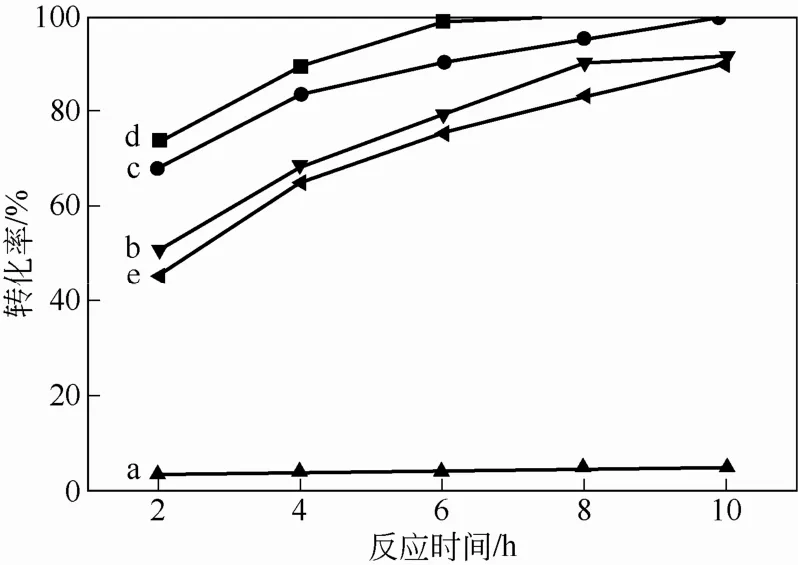
图7 杂化微球对硫化物噻吩和4,6-DMDBT的催化氧化性能
2.2.4 催化剂重复利用效率
反应结束后,通过过滤将复合微球催化剂从体系中分离出来,用丙酮和水交替洗涤处理后,室温下干燥。接着,处理后的杂化微球用30% H2O2重新溶胀,再次投入循环使用。在40℃下,以DBT为硫化物,考察了催化剂重复使用效率,结果见图8。可以明显看出,重复使用3次,催化剂活性基本 保持不变。随着重复次数增加,催化剂活性开始下降,应该是杂化微球表面活性组分脱落造成的。
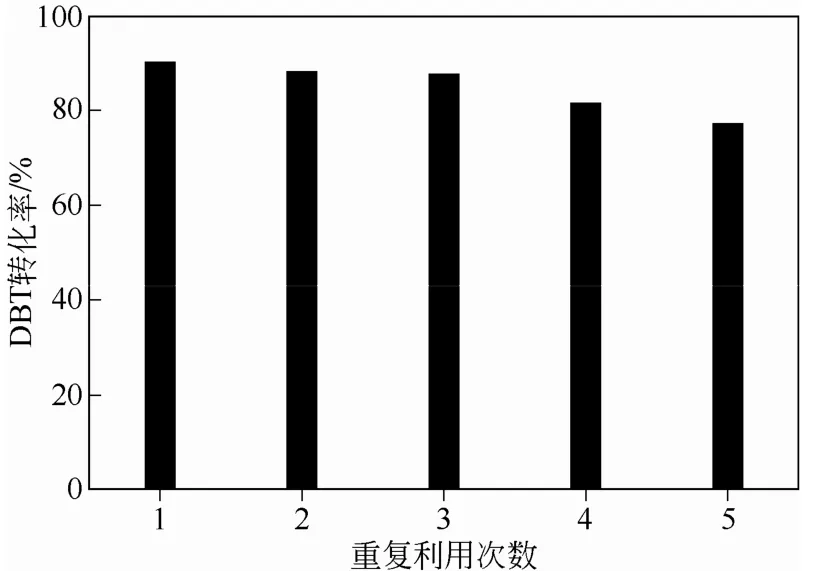
图8 催化剂重复实验效率
2.2.5 催化氧化脱硫机理
基于上述实验表征结果,对PW-HPW/PNIPAM杂化微球作用下、以30% H2O2为氧化剂时硫化物DBT的催化氧化机理进行了探讨,如图9所示。反应起始,在搅拌作用下,杂化微球被均匀分散于反应体系,在杂化微球表面,沉积的杂多酸季胺盐作为相转移催化剂,使得微球内部的H2O2与DBT接触,并继而将DBT氧化为砜。催化氧化反应结束,催化氧化活性成分由过氧磷钨酸季铵盐变为非活性磷钨酸季胺盐。在杂化微球内部的H2O2作用下,非活性磷钨酸季胺盐重新被原位氧化为过氧磷钨酸季胺盐催化剂。催化氧化反应循环反复进行,直至H2O2消耗完全为止。反应结束后,停止搅拌,杂化微球沉积于反应器底部。利用过滤,将杂化微球分离出来,活化后可重复利用。
3 结 论

图9 PW-HPW/PNIPAM杂化微球作用下DBT催化氧化机理示意图
以PNIPAM高聚物微凝胶为模板,成功合成了表面担载有过氧磷钨杂多酸十六烷基季胺盐的杂化微球,并将其作为催化剂用于模拟催化氧化脱硫实验。结果表明,使用过氧化氢为氧化剂,杂化微球不仅对二苯并噻吩具有较高的催化效率,而且对噻吩与4,6-二甲基二苯并噻吩也表现出较强的催化活性。尤为重要的是,该体系为非均相催化氧化体系,杂化微球催化剂能够从体系中进行简易分离,经活化处理后可多次重复利用,而催化性能下降不大。本文仅对杂化微球的催化氧化脱硫性能进行了初步的模拟研究,有关杂化微球在催化深度脱硫方面应用研究的许多细节性工作正在进行之中。但目前实验结果已表明,担载型杂化微球在深度脱硫方面具有独特的优势,这种非均相催化氧化体系不仅有望实现绿色催化氧化深度脱硫,并且对构筑非均相催化微反应器具有普遍的借鉴意义。
[1] US EPA. Regulatory announcement:Heavy-duty engine and vehicle standards and highway diesel fuel sulfur control requirements. EPA420-F-00-057[EB/OL]. 2000. http://www.epa.gov/otaq/diesel. htm.
[2] 李宇慧,冯丽娟,王景刚,等. MoO3/Al2O3-H2O2体系用于柴油催化氧化脱硫[J]. 化工进展,2010,29(s1):659-661.
[3] Yang R T,Hernandez-Maldonado A J,Yang F H. Desulfurization of transportation fuels with zeolites under ambient conditions[J].Science,2003,301:79-81.
[4] Gao X,Du Z,Ding H,et al. Kinetics of NOxabsorption into (NH4)2SO3solution in an ammonia-based wet flue gas desulfurization process[J].Energy Fuels,2010,24(11):5876-5882.
[5] Chapados D,Bonde S E,Gore W L,et al. NPRA AM-00-25[R]. Washington D C:National Petrochemical and Refiners Association,2008.
[6] Filippis P D,Liuzzo G,Scarsella M,et al. Oxidative desulfurization Ⅱ:Temperature dependence of organosulfur compounds oxidation[J].Ind. Eng. Chem. Res.,2011,50(18):10452-10457.
[7] Boron D J,Deever W R,Atlas R M,et al. NPRA AM-99-54[R]. Washington D C:National Petrochemical and Refiners Association,1999.
[8] Vazquez-Duhalt R,Torres E,Valderrama B,et al. Will biochemical catalysis impact the petroleum refining industry?[J].Energy Fuels,2002,16(5):1239-1250.
[9] Collins F M,Lucy A R,Sharp C,et al. Oxidative desulphurisation of oilsviahydrogen peroxide and heteropolyanion catalysis[J].J. Mol. Catal. A:Chem.,1997,117:397-403.
[10] Shao B,Shi L,Meng X. Deep desulfurization of 4,6-dimethyldienzothiophene by an ionic liquids extraction coupled with catalytic oxidation with a molybdic compound[J].Ind. Eng. Chem. Res.,2014,53:6655-6663.
[11] 董阳阳,白金泉,王乔乔. 介孔分子筛固载希夫碱金属配合物催化氧化研究进展[J]. 化学试剂,2013,35(3):233-238.
[12] 姜伟丽,豆丙乾,李沛东,等. 烯烃歧化反应中的负载氧化钨催化剂[J]. 化工进展,2012,31(12):2686-2719.
[13] 宋少飞,周福林,弓巧娟. 气相原位反应制备表面负载纳米银PAM复合微球[J]. 陕西师范大学学报:自然科学版,2013,41(3):36-39.
[14] Yang J X,Fang Y,Bai C L,et al. CuS-poly (N-isopropylacrylamide-co-acrylic acid) composite microspheres with patterned surface structures:Preparation and characterization[J].Chin. Sci. Bull.,2004,49:2026-2032.
[15] Yang J X,Hu D D,Fang Y,et al. Novel method for preparation of structural microspheres poly(n-isopropylacrylamide-co-acrylic acid)/SiO2[J].Chem. Mater.,2006,18:4902-4907.
[16] Wang X J,Hu D D,Yang J X. Synthesis of PAM/TiO2composite microspheres with hierarchical surface morphologies[J].Chem. Mater.,2007,19:2610-2621.
[17] 周美娟,魏长平,毕颖丽,等. 磷钨杂多化合物催化H2O2氧化十八醇制十八酸[J]. 催化学报,1999,20(4):437-441.
[18] 张乾,范晓东. 丙烯酞胺反相微乳液体系的制备、聚合及表征[J].化学工业与工程,2001,18(6):316-340.
[19] 陈小华,陈传盛,孙磊,等. 碳纳米管的表面修饰及其在水中的分散性能研究[J]. 湖南大学学报,2004,31(5):18-21.
[20] Sun Y,Xi Z,Cao G Y. Epoxidation of olefins catalyzed by[π-C5H5NC16H33]3[PW4O16] with molecular oxygen and a recyclable reductant 2-ethylanthrahydroquinone[J].J. Mol. Catal. A:Chemical,2001,166:219-224.
[21] 田雪,刘淑芝,刘英楠,等. 过氧磷钨酸催化氧化柴油深度脱硫实验研究[J]. 化工科技,2012,20(2):17-20.
[22] 宋华,穆金城. 催化氧化脱硫分子筛催化剂研究进展[J]. 化工进展,2011,30(2):303-308.
