顶空气相色谱法测定食用油中溶剂残留的研究
2015-07-25徐清秦国富邹柯婷李永波西安市疾病预防控制中心陕西西安710054
徐清,秦国富,邹柯婷,李永波(西安市疾病预防控制中心,陕西西安710054)
顶空气相色谱法测定食用油中溶剂残留的研究
徐清,秦国富,邹柯婷,李永波*
(西安市疾病预防控制中心,陕西西安710054)
摘要:建立一种顶空气相色谱法测定食用油中的溶剂残留量的方法,并对39份食用菜籽油进行分析测定。采用DB-1701毛细管柱(30 m×0.25 mm×0.25 μm),火焰离子化检测器(FID),载气为氮气;进样口温度200℃;检测器温度250℃;柱温:160℃。80℃平衡30 min,顶空进样,外标法定量。被测物质在1.00 mg/kg~128.00 mg/kg质量浓度范围内呈良好的线性关系,相关系数(r)为0.999 9,在20 mg/kg~60 mg/kg两个浓度水平的加标回收率为99.93%~100.03%,RSD为1.69%~3.79%,检出限为0.01 mg/kg。本方法简单、快速、可靠、灵敏度高,能够满足食用油中残留溶剂的检测分析要求。
关键词:顶空气相色谱;食用植物油;溶剂残留;食品安全
植物油中的残留溶剂是在生产过程中产生的。由于浸出法生产植物油能提高出油率,故其深受植物油生产厂家的青睐,目前我国浸出油生产使用的溶剂是一种混合物,称做六号溶剂,主要成分是正己烷,还有甲基环戊烷、2-甲基戊烷和3-甲基戊烷[1]。溶剂残留量过高,会增加溶剂消耗从而造成经济上的损失,会降低油脂卫生品质[2]。植物油抽提溶剂是由多种烃类所组成的,长期食用,会损害人体中枢神经,使神经细胞内的油脂平衡失调,长期接触会麻醉呼吸中枢,损伤皮肤屏障功能、损害周围神经和造血功能[3]。由于未见本市植物油中残留溶剂报道的资料,且为了了解食品质量安全市场准入制度,实施对我市食用植物油残留溶剂污染的影响,现利用顶空气相色谱法[4-6]将西安市2013年市售食用植物油中残留溶剂含量进行检测并进行分析研究。
1 材料与方法
1.1材料
1.1.1主要仪器与试剂
Agilent G1888-7890A型顶空气相色谱仪:美国安捷伦;PL-L型电子天平:上海民桥医疗器械有限公司;微量进样器:上海医用激光仪器厂;超声清洗器:昆山市超声仪器有限公司;六号溶剂标准品(10 mg/mL):国家粮食局科学研究院;烧杯、滴管。
1.1.2样品
试验所用食用油样品均购自当地农贸市场及超市,所有样品均为未开封桶装油。待分析样品在分析前储存于阴凉干燥地方,打开后留样保存于4℃冰箱。
1.2方法
准确称取10.00 g食用油样品于20 mL顶空瓶中,密封,在顶空加热器中加热30 min后进样测定。
1.3标准曲线的制作
取200 mL一级压榨油,80℃水浴中超声30 min去除干扰,分别准确称取10.00 g去干扰的本底油于洁净干燥的8个顶空瓶中,密封。用200 μL微量进样器通过塞子注入六号溶剂标准液1、2、4、8、16、32、64、128 μL(含量分别为0.01、0.02、0.04、0.08、0.16、0.32、0.64、1.28 mg)。此系列用于标准曲线的测定。
1.4仪器条件
1.4.1气相色谱条件
色谱柱:DB-1701毛细管柱(30 m×0.25 mm× 0.25 μm),进样口温度200℃,载气为高纯氮气,恒流1.5 mL/min,压力21.995 Pa,分流比为1∶1,进样1 μL,柱温160℃,保持6.0 min。氢火焰离子化检测器,检测器温度250℃,H2流量为35 mL/min,空气流量为350 mL/min,尾吹流量20 mL/min。
1.4.2顶空条件
柱箱温度80℃,定量环85℃,传输线90℃,样品瓶平衡30.0 min。
2 结果与分析
2.1毛细管柱、柱温及流速的选择
由于六号溶剂标准含有多种组分,为了计算方便,使各组分在同一时间出峰,本试验准备3根不同极性的色谱柱,分别为中等极性固定相,色谱柱:DB-1701(30 m×0.32 mm×0.25 μm);弱极性固定相,色谱柱:HP-5(30 m×0.32 mm×0.25 μm);极性固定相,色谱柱:DB-WAX(30 m×0.25 mm×0.25 μm)。配制六号溶剂的N,N-二甲基乙酰胺(DMA)在FID检测器上也有响应,但在检测结果中,DMA应与六号溶剂完全分离,且六号溶剂各组分保留时间应一致,所以在试验中需要选择合适的柱温。设定初始柱温为60℃,并逐步使柱温上升到140℃,分别取配制好的标样在相同的顶空条件下,注入到以上色谱柱中,观察标样的出峰情况。
在DB-WAX色谱柱上标样在240℃,流速为2.5 mL/min时六号溶剂各组分保留时间一致且与DMA分离。在HP-5色谱柱上标样在160℃,流速为1.5 mL/min时六号溶剂各组分保留时间一致且与DMA分离,但标准峰有拖尾,如图1所示。
在DB-1701色谱柱上标样在160℃,流速为1.5 mL/min时六号溶剂各组分保留时间一致且与DMA分离良好,峰间距较大且无拖尾现象。
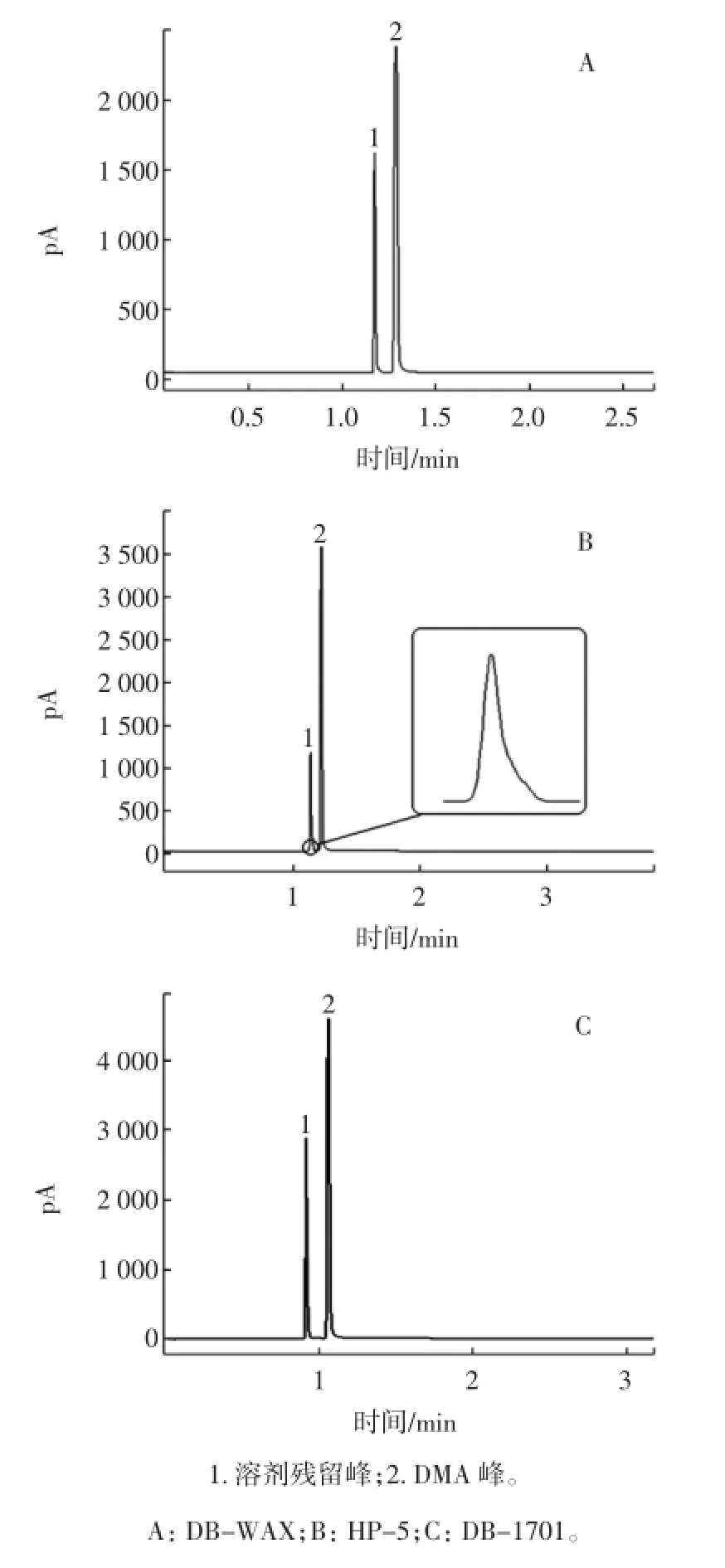
图1 不同极性柱子色谱图Fig.1 Different polarity column chromatography
试验表明:使用中等极性固定相色谱柱DB-1701测定食用油中的溶剂残留比较合适。使用3种色谱柱分离残留溶剂与DMA的色谱条件及检出限如表1所示。
2.2平衡温度对检测结果的影响
分别取配制好的浓度为40 mg/kg的标样在2.1优化的色谱条件下,从40℃起逐步提高平衡体系的温度,每次平衡40 min。随着平衡体系的温度升高,六号溶剂的组分越来越多的挥发到气相中,如图2所示。
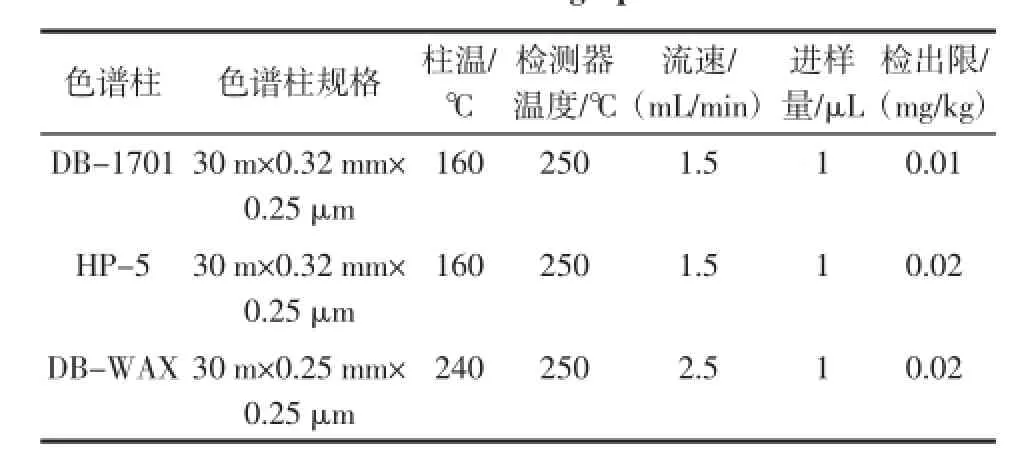
表1 试验所用的3种色谱条件Table 1 Three chromatographic conditions
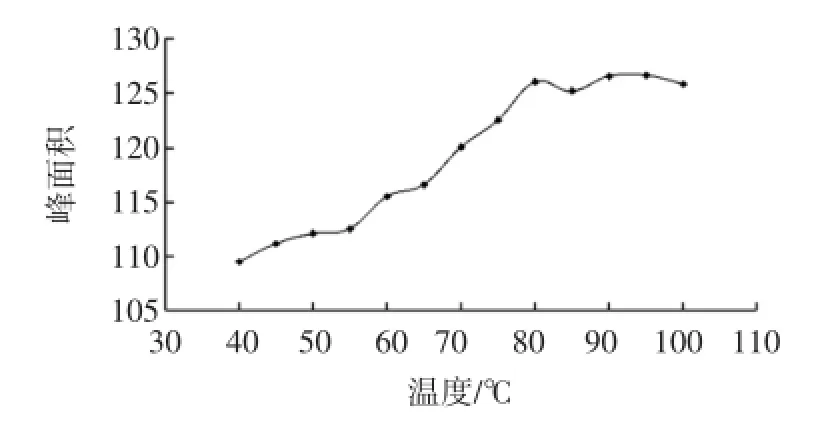
图2 平衡温度对检测结果的影响Fig.2 Affect of equilibrium temperature for test results
在高于80℃平衡温度时,六号溶剂中的大部分组分从本底油中挥发出来,使气液系数达到最大,可检测出挥发烃类的残留溶剂量基本达到最大,从而使检测的灵敏度变大。
2.3平衡时间对检测结果的影响
分别取配制好的浓度为40 mg/kg的标样在2.1与2.2优化的色谱条件下,从10 min起逐步提高平衡时间,平衡温度为80℃。随着平衡时间的逐渐延长,六号溶剂的组分在30 min后基本达到平衡,其含量不再随着平衡时间的增加而增加,如图3所示。

图3 平衡时间对检测结果的影响Fig.3 Effect of equilibrium time for test results
2.4线性范围及检出限
标准曲线如图4所示,由标准曲线可以得出,六号溶剂在1.00 mg/kg~128.00 mg/kg质量浓度范围内呈线性关系良好。以3倍信噪比计算仪器的检出限在0.01 mg/kg。该方法线性范围宽、灵敏度高,可以满足食用油中溶剂残留的分析要求。
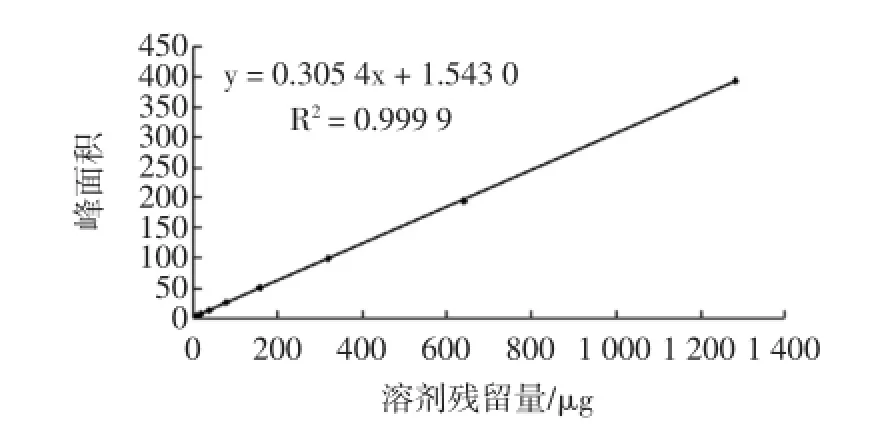
图4 六号溶剂标准曲线Fig.4 VI solvent standard curve
2.5色谱条件
表2列出了本试验与国标检测残留溶剂方法的色谱条件对比。

表2 两种方法检测条件的对比Table 2 Comparison of two methods to detect conditions
2.6回收率和精密度
按照1.2试验方法,分别在去干扰的食用油样品中加入60、20 mg/kg两个水平的标准溶液,重复测定6次,进行加标试验,本底油与加标回收色谱图如图5所示。高低两个水平的加标回收率为分别为100.03% 和99.93%,RSD分别为3.79%和1.69%,详见表3。
2.7实际样品的检测
采用本文建立的方法对在西安农贸市场及超市采集的39份食用油进行了分析测定,有19份检出了残留溶剂。其中浸出油(4级)与压榨油(4级)的检出率为100%,其浓度分别为10.278 mg/kg~34.732 mg/kg和3.551 mg/kg~18.574 mg/kg;浸出油(3级)与压榨油(3级)的检出率分别为88.88%和30%,其浓度分别为1.641 mg/kg~5.697 mg/kg和2.371 mg/kg~10.032 mg/kg;浸出油(1级)、压榨油(1级)与调和油中均未检出残留溶剂,具体结果如表4所示。
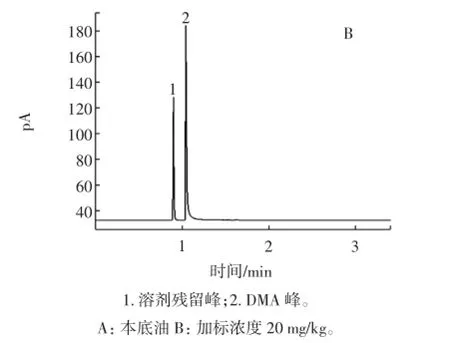
图5 加标样品标准图Fig.5 Standard spiked samples

表3 加标回收率及相对标准偏差Table 3 Recoveries and relative standard deviation

表4 样品中溶剂残留量检出率及检出浓度Table 4 Concentration and positive rate of residual solvent content in edible rapeseed oil

续表4 样品中溶剂残留量检出率及检出浓度Continue table 4 Concentration and positive rate of residual solvent content in edible rapeseed oil
3 结论
为保证食品安全,我国的食用油产品标准GB 1 536-2004《菜籽油》[7]标准对溶剂残留进行了规定。标准规定,采用浸出工艺生产的1、2级植物油中不得检出溶剂残留,3、4级植物油中的溶剂残留量≤50 mg/kg;采用压榨工艺生产的4种级别的植物油中不得检出溶剂残留(注:压榨油和1、2级浸出油的溶剂残留量检出值小于10 mg/kg时,视为未检出)。由标准规定可知,4份浸出油(4级)、1份压榨油(3级)和2份压榨油(4级)检出溶剂残留量。压榨油中检出溶剂残留可能是由于企业生产干燥过程中的加热温度高,会产生烷烃类物质,由于此挥发性烃类物质的存在,检验中会将此烷烃类物质误认为是浸出油的“六号溶剂残留”,从而得出压榨食用植物油不合格的结论[8]。
参考文献:
[1]唐双双.色谱柱极性对测定食用油中溶剂残留的影响[J].农业机械,2013(11):51-52
[2]黄韬睿,王鑫.毛细管气相色谱法测定市售食用油残留溶剂的研究[J].生命科学仪器,2012,10(4):45-46
[3]李艳梅.关于食用油脂中残留溶剂油对人体危害的探讨及对策[J].职业与健康,2001,17(6):46-47
[4]王锡宁,张燕,王艳.毛细管气相色谱法同时测定保健食品中的12中有机溶剂残留量[J].中国卫生检验杂志,2008,18(4):580-581
[5]包志华,张文慧.顶空毛细管柱气相色谱法自动测定食用植物油中残留溶剂[J].内蒙古农业大学学报,2012,33(4):140-142
[6]石允生,李敬章.气相色谱法测定食用植物油中残留溶剂的研究[J].现代预防医学,2005,32(7):787-788
[7]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会.GB1536-2004菜籽油[S].北京:中国标准出版社,2004
[8]张凤梅,郭晓霖.压榨食用植物油溶剂残留量超标分析[J].农产品加工,2011(12):73-74
DOI:10.3969/j.issn.1005-6521.2015.23.037
收稿日期:2014-05-15
作者简介:徐清(1986—),女(汉),副主任技师,本科,研究方向:食品卫生检验。
*通信作者:李永波,女,高级工程师,研究方向:食品安全监测和药物分析。
Study on Methods of Residual Solvent Content in Edible Rapeseed Oil by Headspace Gas Chromatography
XU Qing,QIN Guo-fu,ZOU Ke-ting,LI Yong-bo*
(Xi'an Center For Disease Control and Prevention,Xi'an 710054,Shaanxi,China)
Abstract:A method for determination of residual solvent content in edible rapeseed oil by headspace gas chromatography was established,and 39 rapeseed oil samples were detected.DB-1701 capillary column(30 m× 0.25 mm×0.25 μm)was used for separation,a flame ionization detector(FID),the carrier gas:nitrogen;inlet temperature:200℃;detector temperature:250℃;column temperature:160℃;balance 30 min at 80℃,headspace,quantitative by external standard.There was a good linearity between the peak area and the concentration of VI solvent within 1.0 mg/kg-128.0 mg/kg,the correlation coefficients were 0.999 9,the recoveries of spiked samples at 20 mg/kg-60 mg/kg were ranged from 99.93%-100.03%,RSDs were ranged from 1.69%-3.79%,and the detection limits was 0.01 mg/kg.The method was simple,rapid and reliable.It could be applied to determine residual solvent content in edible rapeseed oil.
Key words:headspace gas chromatography;edible vegetable oils;residual solvent;food safety
