SO42-/MxOy型固体超强酸酸中心结构及其在轻质烷烃异构化反应中的机理研究进展
2015-07-25孟瑶李锋宋华
孟瑶,李锋,2,宋华,2
(1 东北石油大学化学化工学院,黑龙江 大庆 163318;2 东北石油大学石油与天然气化工省重点实验室, 黑龙江 大庆 163318)
烷烃异构化是提高汽油辛烷值的有效手段。早期用于烷烃异构化的酸催化剂是液体酸(如H2SO4和HF),虽然酸强度高,但因其腐蚀设备和污染环境,研究和发展新的环境友好的酸催化剂受到人们广泛关注。固体酸催化剂(包括沸石、复合氧化物、杂多酸等)作为一种新型催化材料,克服了液体酸易腐蚀设备、污染环境、难分离等诸多弊端,成为近年来人们研究的热点[1]。在众多固体酸催化剂中,SO42-/MxOy型固体超强酸因具有催化活性高、选择性好、制备方法简单、不污染环境、不腐蚀设备等突出优点而备受青睐[2]。固体超强酸中研究最多的是SO42-/ZrO2[3],但迄今对其表面结构认识不够深入,对其轻质烷烃异构化反应机理存在一定争议。制备具有高活性、稳定性及反应选择性的SO42-/MxOy型固体超强酸催化剂需要催化剂结构特点及异构化反应机理等深层次的理论研究成果为指导。本文主要介绍了SO42-/MxOy型固体超强酸催化剂表面特征及其酸中心模型,重点阐述了SO42-/MxOy型固体超强酸催化剂对轻质烷烃异构化反应机理及正丁烷异构化反应机理,为制备高性能的SO42-/MxOy型固体超强酸催化剂提供理论指导。
1 SO42-/MxOy 型固体超强酸催化剂的表面特征
SO42-/MxOy型固体超强酸催化剂的制备对氧化物(MxOy)有特殊的要求,并不是所有的金属氧化物都能与SO42-合成固体超强酸。金属离子的电负性和配位数与促进剂SO42-的配位结构有密切关系。此外,固体超强酸的酸催化活性还与金属氧化物的晶态和电子结构有关。
SO42-/MxOy型固体酸催化剂表面的S 具有高氧化态和类似有机硫酸盐结构的S=O 共价键,并通过S—O—M 键以单配位(Ⅰ)、双齿鳌合(Ⅱ)或桥式配位(Ⅲ)状态结合在金属氧化物表面,如图1。红外光谱(IR)分析表明,这3 种结构能在固体表 面产生较强的L 酸或B 酸中心。

图1 金属氧化物与促进剂配位图
SO42-/ZrO2在烷烃异构化反应中具有良好的低温活性,为了说明SO42-/ZrO2表面的活性位性质,人们对其表面结构进行了广泛的研究。然而,目前为止并没有一致的结论。Jin 等[4]通过光电子能谱和红外光谱研究提出了SO42-/ZrO2是一种螯合双齿配合物,硫物种与一个Zr 原子螯合在一起,见图2(a)。Ward 和Ko[5]通过原位和非原位漫反射红外傅里叶变换和X 射线衍射技术研究提出了一个相似的模型,见图2(b)。在这种情况下,羟基与一个Zr 相连接,这个Zr 与一个和硫物种相螯合的Zr 毗邻。由于硫酸物种中两个S=O 的电子诱导效应,质子酸强度增强。Saur 等[6]认为硫酸基团中3 个氧原子和Zr 是以三齿形式连接在一起的,见图2(c)。在H2O存在的条件下,硫酸物种就会转变成桥式双配位结构,同时形成 B 酸中心。Saur 等也提出如果SO42-/ZrO2具有较高的硫含量,多硫结构[图2(d)]就有可能形成,这与Morterra 等[7]的观点相一致。Laizet 等[8]发现当硫含量较低时,硫酸根基团中4个氧原子与锆相接,形成四齿模型,见图2(e)。Rosenberg 和Anderson[9]对SO42-/SiO2-ZrO2进行研究发现,图2(g)结构通过水合作用会生成图2(f)结构。Kustov 等[10]通过漫反射红外光谱对SO42-/ZrO2研究发现,硫酸改性能够增大L 酸和B 酸酸强度,其B 酸模型见图2(h)和图2(i),L 酸模型见图2(j)。Riemer 等[11]提出的另一种结构是两个O 与Zr 以桥式双配位形式连接的模型。有关SO3物种与ZrO2配合的结构模型有两个:其中一个模型结构[12]是SO3中一个O 与一个Zr 连接,SO3中的S 与ZrO2孤对电子相连接,见图2(k);另一个结构模型[13]是SO3中两个O 与ZrO2连接,剩余的S=O 单独存在,见图2(l)。
2 SO42-/MxOy 型固体酸催化剂的酸中心形成机理与调控
SO42-/MxOy型固体超强酸催化剂的酸性中心是影响其催化活性的关键因素。固体酸的超强酸性是由于新生成的或已经存在的B 酸中心产生,其酸中心形成机理如图3 所示。SO42-在催化剂表面吸附配位,由于S=O 的诱导效应,使相应的金属离子得电子能力增强,使M-O 的电子云偏移,从而促进了L 酸中心(Lewis 酸)的形成。L 酸中心吸附水分子后,对水分子中的电子存在强吸引作用,使水分子在干燥和焙烧过程中发生解离、吸附形成B 酸中心。在金属-酸双功能催化剂上的正构烷烃临氢异构化反应中[14],氢气溢流可以提高催化剂表面B 酸酸量。具有加氢-脱氢活性的金属组分一般选自元素周期表中Ⅷ族和ⅥB 族元素,可分为贵金属和非贵金属,贵金属以铂和钯等为主,多以金属单质形式使用;非贵金属主要有钼、镍、钴和钨等。这些金属多以相互结合的硫化物形态使用,这种结构能够提高催化剂的活性和稳定性。B 酸形成过程如图4 所示,吸附在金属Pt 表面的氢气解离成两个氢原子并溢流到SO42--ZrO2表面,然后一个氢原子迁移到L酸中心并给予其电子,氢原子成为B 酸中心,而另一个氢原子接受L 酸中心上的电子形成H--L 酸位。Triwahyono 等[15]通过吸附吡啶红外研究发现,质子酸还可以来源于正戊烷分子。当温度大于350K 时, Pt/SO42--ZrO2催化剂与正戊烷蒸气接触,正戊烷分子解离成一个氢原子和一个戊基自由基。随后氢原子在载体上溢流转移到L 酸中心,并给予L 酸中心一个电子,L 酸中心变成质子酸。所以,在L 酸中心存在的条件下,只要某分子能够在催化剂表面形成氢原子,该分子就能促进B 酸中心的形成[4]。
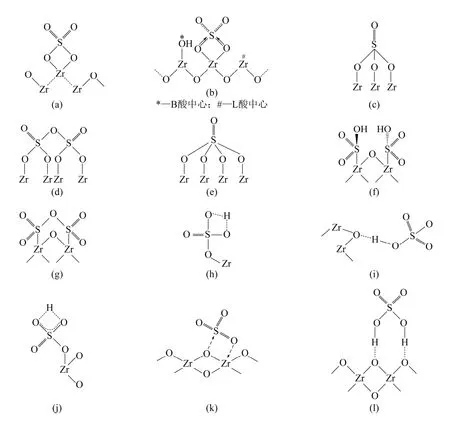
图2 SO42-/ZrO2 表面模型

图3 SO42-/MxOy 型固体超强酸催化剂中的B 酸中心和 L 酸中心形成机理

图4 Pt/SO42--ZrO2 固体酸催化剂上B 酸形成机理
固体超强酸催化剂的酸强度可以通过Hammett指示剂法。表1 总结了不同SO42-/MxOy型固体超强酸催化剂Hammett 指示剂法测得的酸强度数据[3]。由表1 可知,SO42-/SnO2催化剂的酸强度最强,SO42-/ZrO2催化剂次之。1979 年Hino 等[16]首次合成了SO42-/ZrO2固体超强酸催化剂,提出其酸强度超过100%浓硫酸一万倍。Katada 等[17]用NH3-TPD法测出SO42-/ZrO2催化剂的Ho=-19.1,证明了SO42-/ZrO2催化剂具有超强酸性。催化剂中B 酸和L 酸酸量大小,可以通过吡啶或CO 吸附红外分析来测定。不同SO42-/MxOy型固体超强酸催化剂中B酸中心和L 酸中心含量分布不同。Volkova 等[18]由 硝酸氧锆和硝酸铝制备Al2O3/ZrO2时,通过控制氨水的滴加量将溶液pH 值调至7、10 和11.5,并将得到的锆-铝氢氧化物作为载体制备了Pt/SO42-/ Al2O3/ZrO2(用PSZA-x 表示,x 指所用溶液的pH值),其B 酸和L 酸中心含量如表2 所示。制备过程中溶液的pH 值低,有利于B 酸中心的生成,而pH 值高则促进催化剂的L 酸中心生成,且总酸量高。Ce2O3、Yb2O3和La2O3促进的PSZA 固体超强酸催化剂(分别用PSZAC、PSZAY 和PSZAL 表示)的B 酸和L 酸含量如表2 所示。由表2 可知,稀土氧化物的加入可以使催化剂总酸量增大,且B酸的增长量大于L 酸的增长量[19]。Pérez 等[20]制备了一系列不同 Ni 质量分数(1%~9.6%)的Ni/ZrO2-SO42-固体超强酸催化剂(简称xNiZrS,其中x%指催化剂中Ni 的质量分数),并考察了Ni含量对催化剂中B 酸中心和L 酸中心分布的影响
(表2)。由表2 可知,Ni 的引入明显提高催化剂的 L 酸量。不同陈化方法制备的 Pt-SO42-/ Al2O3-ZrO2催化剂酸含量分布见表2。加入异丙醇后陈化的催化剂(I-PSZA)中B 酸和L 酸含量均大于传统陈化方法制备的催化剂(PSZA),而且L酸增加量大于B 酸增加量。Yu Feng 等[21]制备了一系列SO42-/ZrO2-SiO2(MSCx-m∶n)固体超强酸催化剂,其中Zr/Si 摩尔比x 固定为1∶1,m∶n 为原料中H2SO4/HCl 体积比。m∶n 不同,催化剂的L 酸量和B 酸量不同,见表2。
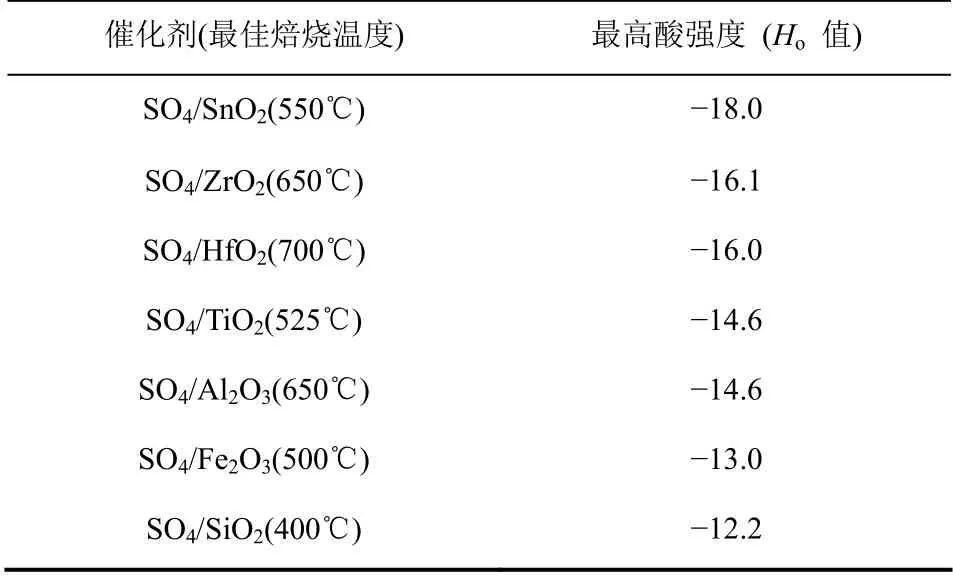
表1 固体超强酸及其酸强度

表2 固体超强酸及其B 酸和L 酸中心含量
本文作者课题组研究了不同温度下焙烧制备的Ni-S2O82-/ZrO2(Ni-SZ-t,t 表示焙烧温度)催化剂的酸含量分布(表2)。随着焙烧温度的提高,B 酸含量逐渐降低,而L 酸含量呈现出先增加后降低的变化趋势;当温度为650℃时,催化剂Ni-SZ-650具有最高的L 酸量和总酸量。综上所述,在固体超强酸催化剂制备过程中,可以通过改变溶液pH 值、引入不同金属元素、加入不同金属含量、改变制备条件等方法调节催化剂表面酸含量,以达到提高其催化活性的目的。
在固体超强酸中,B 酸中心和L 酸中心可以相互转化,Arata[22]研究了SO42-/ZrO2固体超强酸表面B 酸和L 酸转化过程。吸附在SO42-/ZrO2固体超强酸表面的水破坏了S=O 基团与Zr 之间的协同作用,从而产生B 酸中心,且形成的B 酸中心使L酸中心更加稳定。由此,可以提出SO42-/MxOy型固体超强酸催化剂表面B 酸和L 酸相互转化的理论模型,见图5。

图5 L 酸和B 酸相互转化的理论模型
由于SO42-/MxOy型固体超强酸催化剂既含有B酸中心,又含有L 酸中心,两者协同作用下能产生超强酸性,可以代替液体酸催化剂催化异构化反应、烷基化反应、酰化反应、酯化反应和燃烧反应 等[23-26],目前其应用领域已扩展到电化学反应、煤加氢液化反应、光催化反应,燃料电池以及生物柴油方面[27-29]。
在轻质烷烃异构化过程中,SO42-/MxOy型固体超强酸催化剂的催化活性是否仅与B 酸中心或L 酸中心有关,还是与两者都有关系一直有争议。很多学者[30-32]通过实验证明B 酸中心在烷烃异构化过程中是必不可少的,而L 酸并未直接参与到烷烃异构化过程中。Yang 等[33]认为Pt/SZA 对正庚烷的催化活性主要由B 酸起作用,Xu 等[34]发现Fe2O3加入Pt/SO42-/ZrO2后,该催化剂对正庚烷加氢异构化活性相对于Pt/SO42-/ZrO2明显提高,这主要是因为改性后的催化剂中B 酸酸量增加。本文作者课题 组[14,35]在研究Pt 或Pd 改性的催化剂催化正戊烷异构化过程中发现,催化剂中B 酸浓度和酸强度增大,异戊烷收率也相应提高,这证明B 酸在正戊烷异构化过程中起重要作用。B 酸分为弱B 酸、中强B 酸和超强B 酸,不同强度的B 酸在烷烃异构化反应中作用也不同。Hwang 等[36]发现,在Al 和Ga 改性的介孔SZ 固体酸催化剂中,发现强B 酸中心能够促进正丁烷异构化反应的初始反应速率,但也同样会促进积炭,导致原料快速失活;而弱B 酸中心能够保持后续反应阶段的稳定性,所以Hwang 等认为弱B 酸在正丁烷异构化过程中起重要作用。有些学 者[37-38]则支持L 酸中心在烷烃异构化过程中起着重要作用。Volkova 等[18]研究发现在正己烷异构化反应中,L 酸密度越大,正己烷异构化速率越快。这表明催化剂的催化活性与L 酸有关。L 酸也分为弱L 酸、中强L 酸和超强L 酸,不同酸强度的L 酸在烷烃异构化反应中的作用也不同。Yu 等[19]认为在SZ 表面,中强L 酸和超强L 酸对催化剂的催化脱氢异构化活性起着重要作用,中强和超强L 酸酸密度越大,SZ 催化剂的催化性能越好。Hammache 等[39]认为,固体超强酸催化剂对烷烃异构化催化活性与L 酸或B 酸中心密切相关,要想获得高的异构化活性和稳定性,催化剂中L 酸和B 酸中心缺一不可,只有在B 酸中心和L 酸中心协同作用下,催化剂才能发挥更好的催化性能。
3 轻质烷烃异构化反应机理
3.1 轻质烷烃异构化反应机理
轻质烷烃异构化反应机理因催化剂的不同而有所差异,目前国内外学者对烷烃异构化反应机理的解释主要有碳正离子机理、单分子反应机理、双分子反应机理、金属/酸性双功能催化剂催化机理。
(1)碳正离子机理 正构烷烃吸附于催化剂的酸中心上形成碳正离子,形成的碳正离子β 位C—C 键断裂,并使断裂的基团接在碳正离子位上

图6 碳正离子骨架异构化机理

图7 碳离子分解成碳正离子
形成异构碳离子,异构碳离子再与正构烷烃反应生成异构烷烃,如图6 所示。
碳正离子极其活泼,只能瞬时存在,一旦形成就会迅速进行异构化反应。碳正离子机理中的异构化反应可以通过两条途径来实现[40]:A 型异构化和B 型异构化,如图8 所示。A 型异构化包括氢转移和烷基迁移,此反应不改变烷烃的支链度,只改变支链在分子链中的位置,反应速度快。而B 型异构化则为质子角-角迁移,通过质子化环烷碳正离子中间体来增加烷烃的支链度。
(2)单分子反应机理 传统的单分子反应机理认为正构烷烃首先在金属中心上脱氢生成正构烯烃,正构烯烃再在酸性位上质子化为相应的正构烷烃离子,再发生骨架异构化,生成异构烯烃,异构烯烃在金属中心上加氢生成异构烷烃[41]。其反应过程如图9 所示。
正丁烷在固体超强酸催化剂上异构化反应的单分子反应机理模型如图10 所示[42]。
(3)双分子反应机理 双分子反应机理认为,首先烷烃在B 酸中心加氢质子化再脱除一个氢分子形成碳正离子,烷烃在金属位上脱氢形成烯烃。随后,碳正离子与烯烃反应生成二聚体碳正离子,二聚体碳正离子经过重排、β 断裂裂解形成异构烯烃。最后异构烯烃转移到金属位上加氢得到异构烷烃。同时,还会发生歧化反应生成其他副产物。正丁烷在固体超强酸催化剂上异构化反应的双分子反应机理模型如图11 所示[42]。
单分子反应机理和双分子反应机理都是基于碳正离子链传递完成的。通过碳正离子的链式传递方式,异构化反应就可不断进行下去。
(4)金属/酸双功能催化剂催化机理 金属/酸双功能催化剂是目前人们研究的热点,多用于烷烃临氢异构化反应[43-45]。烷烃在金属/酸双功能催化剂上存在单分子反应机理和双分子反应机理,绝大多数反应以单分子反应机理为主。图12 是正庚烷在Pt-WO3/In2O3-ZrO2固体超强酸催化剂上的临氢异构化反应机理[46]。氢分子在反应过程中吸附在还原态的铂粒子上离解成活性氢物种(即H+和H-),n-C7在铂粒子上脱氢形成n-C7=。随后,从H+上得到质子成为n-C7+,n-C7+迅速异构化。一部分i-C7+与H-作用生成 i-C7,一部分 i-C7+裂解成副产物。SO42-/MxOy型金属/酸双功能固体超强酸催化剂催化机理与之类似,如图13 所示。

图8 碳正离子A 型异构化途径和B 型异构化途径
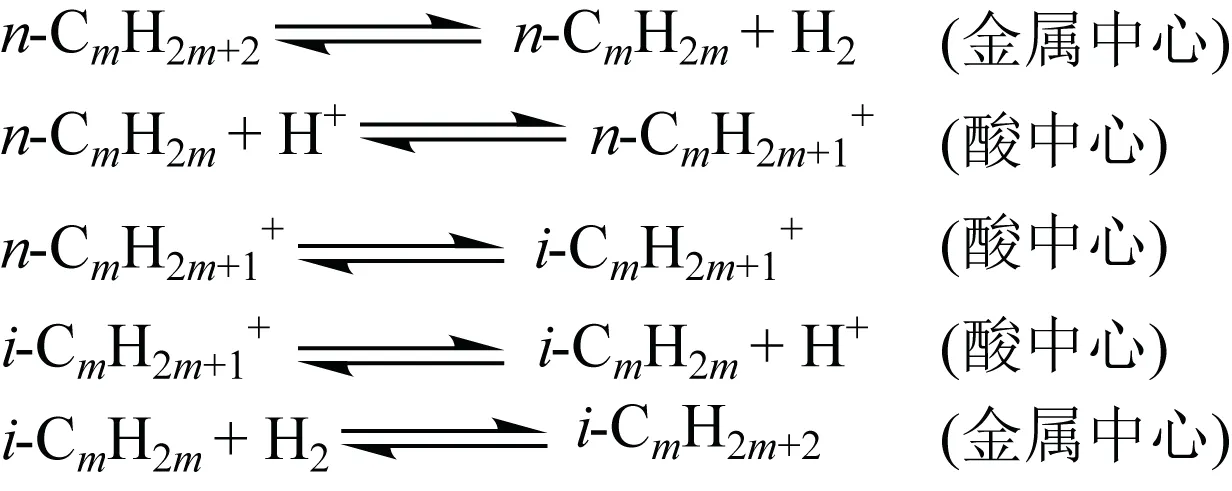
图9 正构烷烃异构化单分子反应机理

图10 正丁烷异构化单分子反应机理
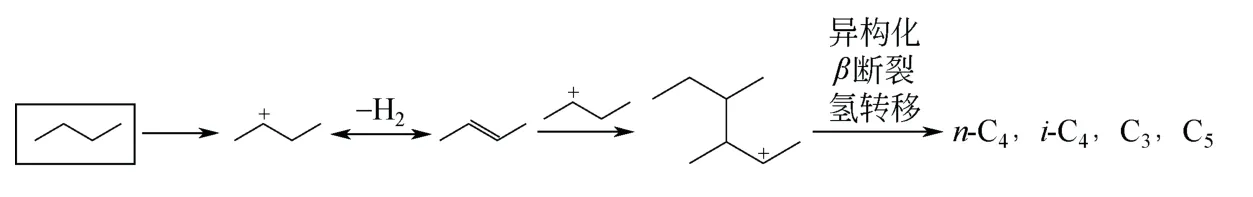
图11 正丁烷异构化双分子反应机理

图12 正庚烷在Pt-WO3/In2O3-ZrO2 上的临氢异构化 反应机理

图13 金属/酸性双功能催化剂催化机理
对于金属/酸双功能催化剂,其异构化反应是金属中心和酸性中心协同催化作用的结果,不能单独完成异构化反应。在烷烃异构化过程中,催化剂的金属中心除了正构烷烃脱氢和异构烯烃加氢的作用外,第二个作用是在临氢条件下金属中心吸附氢分子,并将氢分子离解为H+和H-,H+使B 酸中心恢复。H-稳定叔碳正离子生成异构烷烃,减少二次裂解,提高异构化选择性。
3.2 正丁烷在SO42-/MxOy 型固体酸催化剂上的 异构化反应机理
对于SO42-/MxOy型以及改性的SO42-/MxOy型固体酸催化剂上的正丁烷异构化反应机理,目前主要存在3 种不同的观点:①单分子反应机理;②双分子反应机理;③单分子反应机理和双分子反应机理之间的转变。Ahmed[47]发现,在HfO2改性的SZ固体超强酸催化剂催化的正丁烷异构化反应中,正丁烷直接转变成异丁烷、丙烷和正戊烷。而丙烷和正戊烷的生成是双分子反应机理起作用的明显迹象,这表明该反应以双分子反应机理为主。Adeeva等[48]在研究SZ 和SO42-/Fe2O3-MnO2-ZnO2催化剂对正丁烷的异构化活性时发现,正丁烷异构化也以双分子反应机理进行。Zarkalis 等[49]的动力学研究表明,正丁烷异构化反应的速率方程与可逆二级表面反应相对应,这与双分子反应机理一致。由于正丁烷单分子异构化反应需要经过能垒较高的伯碳离子,而双分子异构化反应需要能垒较低的仲碳离子和叔碳离子,所以双分子反应在能量上是比较有利的[50]。但单分子反应模型副产物少,反应过程中碳效率高,这正是人们所希望的[51]。而且单分子反应机理能够很好地解释在反应初期和较低转化率时催化剂对异丁烷高的选择性。Garin 等[52]研究了1-13C-正丁烷和1-13C-异丁烷在SZ 上的异构化反应,发现当转化率低于30%时,反应以单分子反应机理为主。
以上研究者[46-51]认为丁基碳正离子在催化剂上按单分子机理生成异丁烷或者按双分子机理进行,但也有研究者[53]认为在丁基碳正离子的异构化过程中存在单、双分子机理的转换。Echizen 等[54]研究了1,4-13C 正丁烷在SZ 上的异构化反应,发现在低温时反应以双分子反应机理为主,高温时双分子反应机理和单分子反应机理并存。张丽[55]利用原位固体核磁共振技术研究了正丁烷在SZ 的异构化反应机理,发现反应初期的产物主要为2-13C-正丁烷和1-13C-正丁烷,副产物13C-丙烷和13C-异戊烷主要在反应后期出现,且反应温度的升高导致丙烷和异戊烷生成量的增加。H2的存在可抑制各产物,尤其是副产物的生成。因此,反应初期SZ 上2-13C-异丁烷重构化反应以单分子机理为主,之后向双分子机理转变。这表明氢气气氛有利于反应以单分子机理进行,温度升高有利于正丁烷异构化反应由单分子机理向双分子机理转变。总之,单分子反应机理中,正丁烷分子首先生成丁基碳正离子,丁基碳正离子再经质子化的甲基环丙烷中间体生成异丁烷。单分子反应机理可较好地解释反应初期催化剂对异丁烷的高选择性;正丁烷异构化双分子反应机理中主要涉及由丁基碳正离子和丁烯通过烷基化反应生成C8中间体,C8中间体直接或经异构化反应通过β-断裂生成异丁烷及丙烷和异戊烷等。此机理可以较好地解释反应中副产物的生成及催化剂失活是由积炭导致的。
4 展 望
SO42-/MxOy型固体超强酸催化剂具有较强的酸性、热稳定性,且容易制备与保存,对异构化反应具有良好的活性,是环境友好的新型烷烃异构化催化材料。SO42-/MxOy型固体超强酸及其改性催化剂因其表面结构、酸含量、酸强度不同,其表面上轻质烷烃异构化途径也不同。今后,对于该类催化剂的研究应该注重:①采用原位固体核磁共振等新技术对其表面轻质烷烃异构化机理、活性位形成及失活等方面机理进行深入研究;②对催化剂中促进剂、载体等进行改性,以期获得更好的异构化性能;③探索温和、简洁、高效的制备方法,提高催化剂活性和稳定性;④加强研究解决催化剂与产物的分离、回收、再生以及固体超强酸催化剂制备方法的标准化和系列化等问题,为其工业化提供必要条件。总之,随着人们环保意识的不断增强和对固体酸催化剂的深入研究,SO42-/MxOy型固体超强酸催化剂将作为新型环保催化材料在异构化反应过程中发挥重要作用。
[1] Brown A,Hargreaves J. Methane combustion in the presence of zirconia-based catalysts[J]. Topics in Catalysis,2009,52(5):458-463.
[2] 马千里,所艳华,孟凡龙,等. SO42-/MxOy型固体超强酸催化剂改性及应用研究进展[J]. 工业催化,2014,22(2):85-91.
[3] Arata K,Matsuhashi H,Hino M,et al. Synthesis of solid superacids and their activities for reactions of alkanes[J]. Catalysis Today,2003,81(1):17-30.
[4] Jin Tuo,Yamaguchi Tsutomu,Tanabe Kozo. Mechanism of acidity generation on sulfur-promoted metal oxides[J]. J. Phys. Chem.,1986,90(20):4794-4796.
[5] Ward David A,Ko Edmond I. One-step synthesis and characterization of zirconia-sulfate aerogels as solid superacids[J]. Journal of Catalysis,1994,150(1):18-33.
[6] Saur O,Bensitel M,Saad A B Mohammed,et al. The structure and stability of sulfated alumina and titania[J]. Journal of Catalysis,1986,99(1):104-110.
[7] Morterra C,Cerrato G,Pinna F,et al. Brösted acidity of a superacid sulfate-doped ZrO2system[J]. The Journal of Chemical Physics,1994,98(47):12373-12381.
[8] Laizet J B,Søiland A K,Leglise J,et al. Influence of sulfation and structure of zirconia on catalytic isomerization of n-hexane[J]. Topics in Catalysis,2000,10(1-2):89-97.
[9] Rosenberg Daniel J,Anderson James A. On determination of acid site densities on sulfated oxides[J]. Catalysis Letters,2002,83(1-2):59-63.
[10] Kustov L M,Kazansky V B,Figueras F,et al. Investigation of the acidic properties of ZrO2modified by SO42-anions[J]. Journal of Catalysis,1994,150(1):143-149.
[11] Riemer Thomas,Spielbauer Dieter,Hunger Michael,et al. Superacid properties of sulfated zirconia as measured by Raman and1H MAS NMR spectroscopy[J]. Journal of the Chemical Society,Chemical Communications,1994,10:1181-1182.
[12] Babou F,Bigot B,Coudurier G,et al. Sulfated zirconia for n-butane isomerhation experimental and theoretical approaches[J]. Studies in Surface Science and Catalysis,1994,90:519-529.
[13] White R L,Sikabwe E C,Coelho M A,et al. Potential role of penta-coordinated sulfur in the acid site structure of sulfate zirconia[J]. Journal of Catalysis,1995,157(2):755-758.
[14] 宋华,宋华林,崔雪涵,等. Pd 含量对SO42-/ZrO2-WO3固体超强酸催化剂其异构化性能的影响[J]. 燃料化学学报,2012,40(11):1346-1352.
[15] Triwahyono Sugeng,Jalil Aishah Abdul,Musthofa Malik. Generation of protonic acid sites from pentane on the surfaces of Pt/SO42--ZrO2and Zn/H-ZSM5 evidenced by IR study of adsorbed pyridine[J]. Applied Catalysis A:General,2010,372(1):90-93.
[16] Hino M,Arata K. Solid catalyst treated with anion.1.Catalytic activity of iron oxide treated with sulfate ion for dehydration of 2-propanol and ethanol and polymerization of isobutyl vinyl ether[J]. Chem. Lett.,1979,8(5):477-480.
[17] Katada Naonobu,Endo Jun-ichi,Notsu Kei-ichi. Superacidity and catalytic activity of sulfated zirconia[J]. J. Phys. Chem. B,2000,104(44):10321-10328.
[18] Volkova G G,Reshetnikov S I,Shkuratova L N. n-Hexane skeletal isomerization over sulfated zirconia catalysts with different Lewis acidity[J]. Chemical Engineering Journal,2007,134(1-3):106-110.
[19] Yu G X,Lin D L,Hu Y,et al. RE2O3-promoted Pt-SO42-/ZrO2-Al2O3catalyst in n-hexane hydroisomerization[J]. Catalysis Today,2011,166(1):84-90.
[20] Pérez M,Armendáriz H,Toledo J A,et al. Preparation of Ni/ZrO2-SO42-catalysts by incipient wetness method:Effect of nickel on the isomerization of n-butane[J]. Journal of Molecular Catalysis A:Chemical,1999,149:169-178.
[21] Yu Feng,Guo Min,Wang Xu,et al. Synthesis of well-ordered SO42-/ZrO2-SiO2materials in bi-acid system[J]. J. Fuel Chem. Technol.,2013,41(4):456-462.
[22] Arata Kazushi. Organic syntheses catalyzed by superacidic metal oxides : Sulfated zirconia and related compounds[J]. Green Chemistry,2009,11:1719-1728.
[23] Testa Maria Luisa,Parola Valeria La,Liotta Leonarda F,et al. Screening of different solid acid catalysts for glycerol acetylation[J]. Journal of Molecular Catalysis A:Chemical,2013,367:69-76.
[24] Kuwahara Yasutaka,Kaburagi Wako,Nemoto Koji,et al. Esterification of levulinic acid with ethanol over sulfated Si-doped ZrO2solid acid catalyst : Study of the structure-activity relationships[J]. Applied Catalysis A:General,2014,476(22):186-196.
[25] 李三喜,徐妍如,王松. SO42-/TiO2-HZSM-5 固体超强酸催化剂的制备及酯化性能[J]. 化工进展,2015,34(3):745-750.
[26] Kinnunen Niko M,Hirvi Janne T,Venalainen Tapani,et al. Procedure to tailor activity of methane combustion catalyst:Relation between Pd/PdOxactive sites and methane oxidation activity[J]. Applied Catalysis A:General,2011,397(1-2):54-61.
[27] Gao Yilong,Wu Jianxiang,Zhang Wei,et al. SO42-/SnO2as a new electrode for electrochemical[J]. Ceramics International,2014,40(6):8925-8929.
[28] Iannaci Alessandro,Mecheri Barbara,D’Epifanio Alessandra,et al. Sulfated zirconium oxide as electrode and electrolyte additive for direct methanol fuel cell applications[J]. International Journal of Hydrogen Energy,2014,39(21):11241-11249.
[29] Alhassan Fatah H,Rashid Umer,Taufiq-Yap Y H. Synthesis of waste cooking oil-based biodiesel via effectual recyclable bi-functional Fe2O3-MnO-SO42-/ZrO2nanoparticle solid catalyst[J]. Fuel,2015,142(15):38-45.
[30] Li Xuebing,Nagaoka Katsutoshi,Lercher Johannes A. Labile sulfates as key components in active sulfated zirconia for n-butane isomerization at low temperatures[J]. Journal of Catalysis,2004,227:130-137.
[31] Wang Jung-Hui,Mou Chung-Yuan. Alumina-promoted mesoporous sulfated zirconia:A catalyst for n-butane isomerization[J]. Applied Catalysis A:General,2005,286:128-136.
[32] Song Hua,Wang Na,Song Hualin,et al. La-Ni modified S2O82-/ZrO2-Al2O3catalyst in n-pentane hydroisomerization[J]. Catalysis Communications,2015,59:61-64.
[33] Yang Ying-Chieh,Weng Hung-Shan. Al-promoted Pt/SO42-/ZrO2with low sulfate content for n-heptane isomerization[J]. Applied Catalysis A:General,2010,384(1-2):94-100.
[34] Xu Xin,Liu Tian,Xie Pengfei. Enhanced catalytic performance over Fe2O3-doped Pt/SO42-/ZrO2in n-heptane hydroisomerization[J]. Catalysis Communications,2014,54:77-80.
[35] 宋华,董鹏飞,石洋.Pt 含量及活化温度对固体超强酸催化剂异构化性能的影响[J].高等学校化学学报,2011,32(2):355-360.
[36] Hwang Chi-Chau,Mou Chung-Yuan. Comparison of the promotion effects on sulfated mesoporous zirconia catalysts achieved by alumina and gallium[J]. Applied Catalysis A:General,2009,365(2):173-179.
[37] Watanabe Katsuya,Oshio Nobuyasu,Kawakami Takahito,et al. Isomerization reactions with sulfur-containing pentane over Metal/SO42-/ZrO2catalysts[J]. Applied Catalysis A:General,2004,272(1-2):281-287.
[38] Funamoto Takako , Nakagawa Takamasa , Segawa Kohichi. Isomerization of n-butane over sulfated zirconia catalyst under supercritical conditions[J].Applied Catalysis A:General,2005,286(1):79-84.
[39] Hammache Sonia,Jr James G Goodwin. Characteristics of the active sites on sulfated zirconia for n-butane isomerization[J]. Journal of Catalysis,2003,218(2):258-266.
[40] Blomsma E,Martens J A,Jacobs P A. Mechanisms of heptane isomerization on bifunctional Pd/H-beta zeolites[J]. Journal of Catalysis,1996,159(2):323-331.
[41] López Carmen M,Sazo Virginia,Pérez Pedro,et al. n-Pentane hydroisomerization on Pt-promoted acid zeolites[J]. Applied Catalysis A:General,2010,372(1):108-113.
[42] 贺鹤勇,邹艳,马卓娜,等. 低碳烷烃催化反应机理的固体核磁共振研究[J]. 物理化学学报,2004,20(专刊):1204-1031.
[43] Aboul-Gheit A K,Gad F K,Abdel-Aleem G M. Pt,Re and Pt-Re incorporation in sulfated zirconia as catalysts for n-pentane isomerization[J]. Egyptian Journal of Petroleum,2014,23(3). DOI:10.1016/j.ejpe.2014.08.006.
[44] Urzhuntsev G A,Ovchinnikova E V,Chumachenko V A. Isomerization of n-butane over Pd-SO42-/ZrO2catalyst:Prospects for commercial application[J]. Chemical Engineering Journal,2014,238:148-156.
[45] 宋华,孙恩浩,李锋,等.焙烧温度对微乳液法负载铂制备的Pt-S2O82-/ZrO2-Al2O3催化剂异构化性能的影响[J]. 燃料化学学报,2013,41(6):715-721.
[46] Nie Yingying,Shang Shuning,Xu Xin,et al. In2O3-doped Pt/WO3/ZrO2as a novel efficient catalyst for hydroisomerization of n-heptane[J]. Applied Catalysis A:General,2012,433/434:69-74.
[47] Ahmed M A. Surface characterization and catalytic activity of sulfated-hafnia promoted zirconia catalysts for n-butane isomerization[J]. Fuel Processing Technology,2011,92(5):1121-1128.
[48] Adeeva V,Dehaan J W,Janchen J,et al. Acid sites in sulfated and metal-promoted zirconium dioxide catalysts[J]. Journal of Catalysis,1995,151(2):364-372.
[49] Zarkalis A S,Hsu C Y,Gates B C. Solid superacid catalysis-kinetics of butane isomerization catalyzed by a sulfated oxide containing iron,manganese,and zirconium[J]. Catalysis Letters,1994,29(1/2):235-239.
[50] Li Xuebing,Nagaoka Katsutoshi,Simon Laurent J,et al. Mechanism of butane skeletal isomerization on sulfated zirconia[J]. Journal of Catalysis,2005,232(2):456-466.
[51] Essayem N,Taârit Y Ben,Feche C,et al. Comparative study of n-pentane isomerization over solid acid catalysts,heteropolyacid,sulfated zirconia,and mordenite:Dependence on hydrogen and platinum addition[J]. Journal of Catalysis,2003,219(1):97-106.
[52] Garin F,Seyfried L,Girard P,et al. A skeletal rearrangement study of labeled butanes on a solid superacid catalyst :Sulfuric-acid treated zirconium-oxide[J]. Journal of Catalysis,1995,151(1):26-32.
[53] Nattaporn Lohitharn,Edgar Lotero,James G Goodwin Jr,et al. A comprehensive mechanistic pathway for n-butane isomerization on sulfated zirconia[J]. Journal of Catalysis,2006,241(2):328-341.
[54] Echizen Tsuneo,Suzuk Tetsuo,Kamiy Yuichi,et al. Mechanistic study on skeletal isomerization of n-butane using 1,4-13C2-n-butane on typical solid acids and their Pt-promoted bifunctional catalysts[J]. Journal of Molecular Catalysis A:Chemical,2004,209:145-153.
[55] 张丽. 原位固体核磁共振技术研究催化剂酸碱性质及正丁烷异构化反应[D]. 上海:复旦大学,2011.
