一个脊髓小脑共济失调家系的分子遗传学研究
2015-06-28吴丹丹刘小璇刘建元刘勇陶襄萍杨树广樊东升刘少君
吴丹丹,刘小璇,刘建元,刘勇,陶襄萍,杨树广,樊东升,刘少君
一个脊髓小脑共济失调家系的分子遗传学研究
吴丹丹,刘小璇,刘建元,刘勇,陶襄萍,杨树广,樊东升,刘少君
目的研究1个汉族脊髓小脑共济失调(SCAs)家系的基因分型。方法收集该SCAs家系中的6例患者和有血缘关系的40例无症状者共46人的外周血样品,结合先证者的临床体征及二代测序结果,采用多聚酶链式反应(PCR)对该家系致病基因ATXN2的CAG三核苷酸重复片段进行扩增,通过琼脂糖凝胶电泳和PCR产物测序的方法确定该家系所有成员的正常与异常扩增等位基因内三核苷酸重复次数。结果该家系的遗传性SCAs为常染色体显性遗传,4代46人中有6例SCA2患者,7例为致病等位基因携带者。患者ATXN2两个等位基因中的1条CAG三核苷酸重复次数在正常范围内,另1条CAG三核苷酸重复次数在异常范围。患者异常等位基因CAG三核苷酸重复次数范围为40~46次。结论该家系为CAG三核苷酸重复序列动态突变引起的常染色体显性遗传性脊髓小脑共济失调Ⅱ型,基因诊断还发现该家系中有7名致病等位基因携带者。
脊髓小脑共济失调;三核苷酸重复;动态突变;基因诊断
脊髓小脑共济失调(spinocerebellar ataxias,SCAs)是一大类由遗传因素引起的迟发型神经系统退行性病变,占神经系统遗传性疾病的10%~15%,具有高度的临床和遗传异质性[1-2]。该病是由三核苷酸或五核苷酸动态突变扩展引起的,多呈常染色体显性遗传,其病理改变主要是脊髓、小脑和脑干的神经元变性脱失与胶质增生[2-3],临床表现为小脑共济失调伴构音障碍、意向性震颤、锥体系及锥体外系症状等[4]。根据其临床特点与基因定位可分为SCA1-30共30种亚型[5]。该病虽病情进展缓慢,但呈进行性恶化与遗传早现现象,严重危害患者健康,给家庭和社会带来沉重负担。本研究收集到1个甘肃地区汉族遗传性脊髓小脑共济失调家系并进行研究,对先证者进行核磁共振扫描及肌电图检查,结合临床症状,确诊为脊髓小脑共济失调,但亚型无法确定。取先证者及其弟弟的外周血进行外显子测序,获得与该病相关的突变基因ATXN2,应用聚合酶链反应(polymerase chain reaction,PCR)扩增ATXN2基因,结果发现该家系中6例患者及7例致病等位基因携带者的ATXN2基因中包含1条异常等位基因,异常等位基因内的CAG异常扩增,而家系内其他成员ATXN2基因的CAG重复均在正常范围内。
1 资料与方法
1.1 家系调查 本家系收集自甘肃地区,家系成员5代77人,其中5人因该病去世。家系第一代由于年代久远且都已去世,详细资料无法收集。剩余成员中有6例患者,患者均有不同程度的小脑共济失调症状,其他人表现正常。在得到军事医学科学院伦理委员会同意并征得家系成员知情同意的情况下,采集家系成员46人的外周血各4ml,冻存于本实验室超低温冰箱内备用。
1.2 疾病诊断 先证者于2014年年初在甘肃省临夏州人民医院接受核磁共振检查,随后在北京大学第三医院进行了临床体征检查与肌电图检查。
1.3 致病基因检测
1.3.1 外显子捕获及二代测序 将先证者及其弟弟外周血各200µl送华大基因进行外显子测序,测序平台为HiSeq 2000,获得与该家系疾病相关的突变基因。
1.3.2 基因组DNA提取 使用天根生物科技公司的血液基因组DNA提取试剂盒(离心柱型)提取家系成员血液基因组DNA,用作PCR反应模板。
1.3.3 PCR扩增ATXN2基因 对于华大基因外显子测序所获与该家系疾病相关的突变基因ATXN2,使用Primer Premier 5.0设计引物,正向引物为5'-ACGCCACCACAGCCTCCAGAC-3',反向引物为5'-CTCGCCTCCCTCCGCCTCAG-3',送至生工生物工程有限公司合成。PCR反应体系:总体积50µl,TaKaRa PrimeSTAR Max Premix(2×)25µl,上下游引物(20µmol/L)各1µl,基因组DNA 2µl,双蒸水21µl。PCR反应条件:95℃ 5min预变性;98℃ 20s变性、66℃ 15s退火、72℃ 60s延伸,38个循环;72℃ 7min。PCR产物片段大小为965bp。
1.4 致病基因ATXN2内CAG三核苷酸重复次数的确定 对1.3中的PCR产物采用1%琼脂糖凝胶电泳检测,使用天根生物科技公司的琼脂糖凝胶DNA回收试剂盒(普通离心柱型)对目的条带进行切胶回收,将回收后的DNA送至生工生物工程有限公司进行双向测序,比对测序结果,计数CAG三核苷酸重复次数。
2 结 果
2.1 家系分析及临床诊断
2.1.1 家系分析 调查发现该家系共5代77人,在世成员中有6例患者,发病年龄20~40岁,患者均有不同程度的小脑共济失调症状。该家系中无近亲婚配史,5代中均有患者,男女均可患病,患者的双亲之一必定为患者,符合常染色体显性遗传性疾病的遗传特点(图1)。
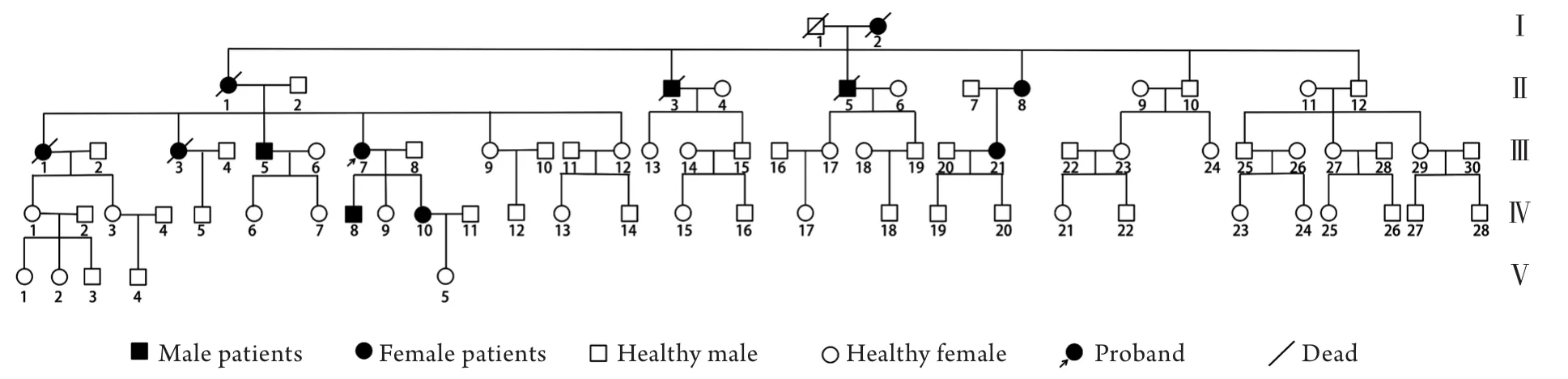
图1 甘肃地区脊髓小脑共济失调家系系谱图Fig.1 The pedigree diagram of the family with spinocerebellar ataxias in Gansu
2.1.2 临床诊断 先证者,Ⅲ-7,38岁发病,发病7年。患者7年前无明显诱因出现行走不稳伴持物不稳,双下肢发僵,进食呛咳,并逐渐加重,其余无异常。临床体征:双上肢震颤,双下肢发抖,行走不稳。神经系统检查:轻度构音障碍,指鼻及跟-膝-胫试验欠稳妥,轮替障碍,辨距不良。肌电图检查:双上肢和下肢深感觉传导路径阻滞,双侧听觉-脑干径路波幅下降,双侧视觉径路波幅下降,四肢肌张力下降,腱反射减弱。头部核磁扫描显示:小脑和脑干萎缩,以小脑萎缩为著(图2)。结合以上资料,确诊先证者患有脊髓小脑共济失调。
2.2 致病基因检测
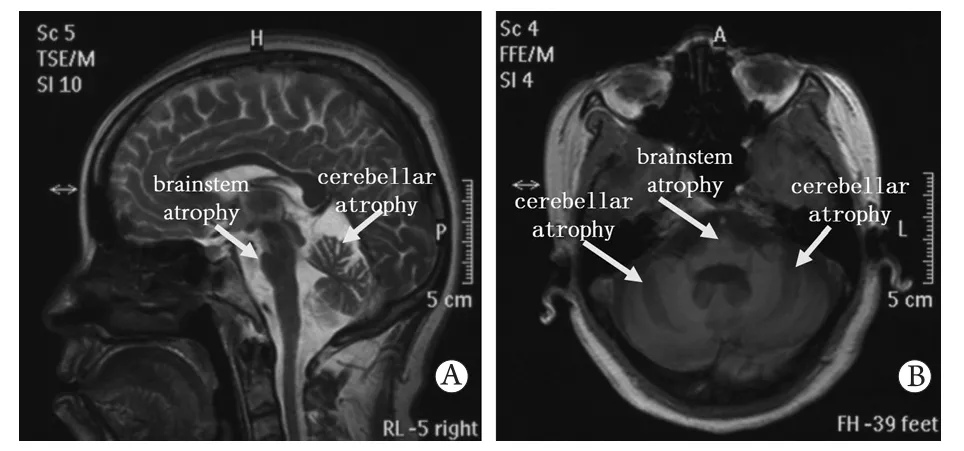
图2 先证者头颅MRI扫描(箭头示意脑萎缩部位)Fig.2 Brain MRI images of proband (arrows point to the brain atrophy) A. Sagittal scanning; B. Axial scanning
2.2.1 外显子捕获及二代测序 在华大基因测序的两位患者外显子测序结果与人类基因组参考序列(UCSC hg 19 version)比对,两位患者相同的突变为84个点突变位点和8个插入缺失突变基因。结合临床诊断结果,首先选取8个插入缺失突变基因中的ATXN2基因进行PCR验证。
2.2.2 PCR产物琼脂糖凝胶电泳检测 对46位家系成员进行了突变基因ATXN2的检测,结果发现家系中的6例患者Ⅱ-8、Ⅲ-5、Ⅲ-7、Ⅲ-21、Ⅳ-8、Ⅳ-10及7例致病等位基因携带者Ⅲ-13、Ⅳ-1、Ⅳ-6、Ⅳ-19、Ⅴ-1、Ⅴ-3、Ⅴ-5的PCR产物电泳图出现明显的2条大小不等的条带,而家系中其他成员未出现该现象。这是因为理论上针对这一位点而言,每个个体PCR产物都是两个条带(一对等位基因),正常人两条等位基因的CAG重复次数相同或者差距较小,在电泳图上无法区分,但是患者和携带者的PCR产物的正常等位基因明显比异常等位基因小,在电泳图上可分辨出来。对PCR扩增后的条带切胶回收DNA,送至生工生物工程有限公司进行双向测序。
2.2.3 比对计数CAG重复序列扩增次数 回收后的DNA片段采取ATXN2双向测序,根据测序结果计数致病基因ATXN2的CAG(反向为CTG)三核苷酸重复次数(图3、4),结果显示患者及致病等位基因携带者异常等位基因的CAG重复次数为40~48次,明显超出正常范围(表1、2),剩余家系成员ATXN2两条等位基因的CAG重复次数均在19~25次,属于正常范围。

表1 患者CAG重复次数Tab. 1 The CAG repeats of patients
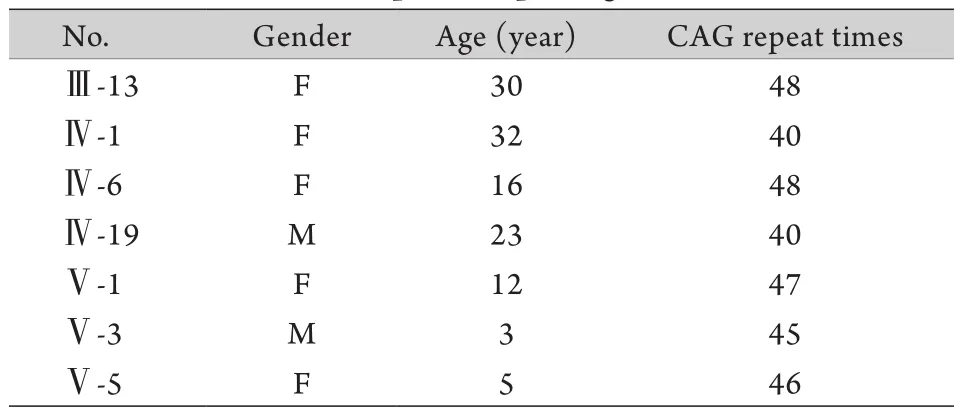
表2 异常等位基因携带者CAG重复次数Tab. 2 The CAG repeats of pathogenic allele carriers

图3 正常对照Ⅱ-10 PCR产物测序结果Fig. 3 Sequencing result of the PCR products of normal control Ⅱ-10

图4 患者Ⅱ-8 PCR产物测序结果Fig. 4 Sequencing result of the PCR products of patient Ⅱ-8
3 讨 论
SCAs是一大类具有高度临床和遗传异质性的神经系统进展性退行性病变,以小脑共济失调,构音障碍,眼球震颤,认知障碍,锥体系及锥体外系、脊髓及外周神经系统受累为特征[6],其主要病理改变是神经元变性脱失和神经纤维的沃勒变性[7]。目前已有30多种SCAs亚型的致病基因被定位,其中SCA1、SCA2、SCA3/MJD、SCA6、SCA7、SCA17及齿状核-红核-苍白球路易体萎缩(dentatorubralpallidoluy-sian atrophy,DRPLA)这7种亚型是由于致病基因编码区内的CAG三核苷酸重复扩增使其编码的多聚谷氨酰胺链异常扩展而引起的[8]。有研究表明,SCAs中的CAG三核苷酸重复扩增可导致中枢神经系统核包涵体形成,有可能是引发SCAs疾病的原因[9]。
SCA2是在古巴最早被发现并描述的,且在古巴的发病率较高[10],在中国则较少见[11]。SCA2患者多在30~40岁发病[12],病程进展缓慢,临床表现以小脑共济失调、构音障碍、眼肌麻痹、慢眼动、腱反射减弱、早期周围神经病变等症状为主,常伴智力障碍,头部MRI检查显示不同程度的橄榄-脑桥-小脑萎缩[13]。SCA2患者的病理损伤主要存在于小脑皮质的浦肯野细胞和脑干神经元,还存在于大脑皮质的额叶和枕叶以及肌肉组织[7]。
ATXN2是SCA2的致病基因,最先被德国的Gispert与Twells在1993年定位于12q23-24.1[14],由Pulst 等[15]在1996年成功克隆。ATXN2基因编码Ataxin-2蛋白,相对分子质量约为140×103,含1312个氨基酸残基,第1外显子编码区CAG三核苷酸异常重复扩展导致编码蛋白中的多聚谷氨酰胺链延长而致病[16]。正常ATXN2等位基因CAG三核苷酸重复次数为13~31次,而异常等位基因重复次数大于34次,多数在36~59次之间,且序列中没有CAA插入[15],中间重复范围为32~34次[17]。
本研究的家系先证者以躯干共济失调起病,随后出现姿势性震颤,运动神经正常,感觉神经的双侧尺神经和一侧腓神经未测出,双侧听觉-脑干径路波幅下降,双侧视觉径路波幅下降,有颅脑神经损伤,无明显慢眼动,无明显周围神经病变,小脑系症状有构音障碍、眼肌麻痹、躯干共济失调、姿势性震颤等,锥体外系症状有铅管样肌张力增高、静止性震颤等。该先证者的症状基本符合SCA2的表现,但其家系成员均无智力障碍。基因诊断方面,患者与正常人CAG三核苷酸重复均被CAA打断,CAA 与CAG一样编码谷氨酰胺,但有报道CAA插入可能会减轻患者的症状[18],这在本研究中有所体现,CAG重复中CAA插入越多的患者症状相对越轻。除此之外,有报道称患者CAG重复次数越多,则发病年龄越小,病情越严重[19],这在本家系成员中没有体现,亦未显出现其他报道中的遗传早现现象。
本研究所搜集到的家系具有以下特点:①家系庞大,血液样本采集较全;②所有患者均无智力障碍,表现为智力正常;③SCA2在该家系的不同成员中具有临床和遗传异质性;④家系中7例SCA2症状前患者异常等位基因CAG重复次数为40~48次,远远超出正常人CAG重复13~31次的范围,可以做出7例为致病等位基因携带者的诊断。本研究除了对ATXN2进行PCR检测验证外,还对外显子测序所获的84个点突变位点和其他7个插入缺失突变基因进行了PCR检测验证,结果证实这些突变基因均与该家系疾病无关,说明对于SCA2,ATXN2基因内CAG三核苷酸异常扩展是发病的主要原因。
本研究对于该家系的遗传性神经系统疾病进行了明确诊断,确诊为脊髓小脑共济失调Ⅱ型,具有重要的理论与现实意义。首先,为家系成员提供了翔实的资料,明确了家系成员携带致病基因的情况,为他们的医疗保健提供了参考。其次,如此庞大的SCA2家系在中国乃至世界范围内都比较少见,该家系成员的临床体征、核磁共振影像、肌电图检查等资料丰富了SCA2的研究。再次,该家系成员的临床体征结合基因诊断结果,再一次证实了SCAs分型不能仅仅依靠临床诊断,因为SCAs具有高度的临床和遗传异质性,且各亚型之间有重叠症状,必须结合基因诊断。
[1]Wu Y, Wei QQ, Shang HF. Frequency analysis and clinical characterization of different types of spinocerebellar ataxia[J]. Chin J Pract Intern Med, 2014, 34(5): 512-515. [吴英, 魏倩倩,商慧芳. 脊髓小脑共济失调基因型分布及临床特点分析[J].中国实用内科杂志, 2014, 34(5): 512-515.]
[2]Liu D, Gou H, Wang K,et al. Genetics of a Chinese family with spinocerebellar ataxia[J]. J Third Mil Med Univ, 2011, 33(11): 1152-1154. [刘丹, 郭洪, 王凯, 等. 脊髓小脑共济失调一家系的遗传学研究[J]. 第三军医大学学报, 2011, 33(11): 1152-1154.]
[3]Gu WH, Wang GX. Diagnosis and differentiation of olive pons cerebellum atrophy, spinocerebellar ataxia, and multiple system atrophy-cerebellar type[J]. Chin J Neurol, 2008, 41(2): 135-137. [顾卫红, 王国相. 橄榄脑桥小脑萎缩、脊髓小脑共济失调和多系统萎缩小脑型的诊断和鉴别[J]. 中华神经科杂志, 2008, 41(2): 135-137.]
[4]Honti V, Vécsei L. Genetic and molecular aspects of spinocerebellar ataxias[J]. Neuropsychiatr Dis Treat, 2005, 1(2): 125-133.
[5]Matilla-Duenas A, Sánchez I, Corral-Juan M,et al. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias[J]. Cerebellum, 2010, 9(2): 148-166.
[6]Manto M, Marrmolino D. Cerebellar ataxias[J]. Curr Opin Neurol, 2009, 22(4): 419-429.
[7]Zhang XN, Wang SZ. The research progress of spinocerebellar ataxia[J]. Xinjiang Med, 2006, 36(5): 206-210. [张小宁, 汪师贞. 脊髓小脑共济失调的研究进展[J]. 新疆医学, 2006, 36(5): 206-210.]
[8]Traoré M, Coulibaly T, Meilleur KG,et al. Clinical and genetic analysis of spinocerebellar ataxia in Mali[J]. Eur J Neurol, 2011, 18(10): 1269-1271.
[9]Mori F, Tanji K, Odagiri S,et al. Autophagy-related proteins (p62, NBR1 and LC3) in intranuclear inclusions in neurodegenerative diseases[J]. Neurosci Lett, 2012, 522(2): 134-138.
[10] Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond[J]. Lancet Neurol, 2010, 9(9): 885-894.
[11] Jiang H, Tang BS. Clinical and genetic diagnosis progress of spinocerebellar ataxia[J]. Foreign Med Sci Neurol Neurosurg, 2002, 29(4): 290-293.[江泓, 唐北沙. 脊髓小脑性共济失调的临床及基因诊断进展[J]. 国外医学神经病学神经外科学分册, 2002, 29(4): 290-293.]
[12] Matilla-Duenas A, Goold R, Giunti P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1[J]. Cerebellum, 2008, 7(2): 106-114.
[13] Li ZC, Tian ZM. Progression in the study of spinocerebellar ataxia[J]. Acad J Sec Mil Med Univ, 2005, 26(6): 692-694. [李志超, 田增民. 脊髓小脑共济失调的研究进展[J]. 第二军医大学学报, 2005, 26(6): 692-694.]
[14] Gispert S, Twells R, Orozco G,et al. Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1[J]. Nat Genet, 1993, 4(3): 295-299.
[15] Pulst SM, Nechiporuk A, Nechiporuk T,et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2[J]. Nat Genet, 1996, 14(3): 269-276.
[16] Chen YY, Hao Y, Gu WH,et al. Clinical and neuroimaging study of spinocerebellar ataxia type 2[J]. Chin Contemp Neuro Neurosurg, 2013, 13(6): 525-532. [陈园园, 郝莹, 顾卫红, 等.脊髓小脑共济失调2型临床和神经影像学特征分析[J]. 中国现代神经疾病杂志, 2013, 13(6): 525-532.]
[17] Laffita-Mesa JM, Velázquez-Pérez LC, Santos Falcón N,et al. Unexpanded and intermediate CAG polymorphisms at SCA2 locus (ATXN2) in the Cuban population: evidence about the origin of expanded SCA2 alleles[J]. Eur J Hum Genet, 2012, 20(1): 41-49.
[18] Matsuyama Z, Izumi Y, Kameyama M,et al. The effect of CAT trinucleotide interruptions on the age at onset of spinocerebellar ataxia type 1 (SCA1)[J]. J Med Genet, 1999, 36(7): 546 -548.
[19] Shu AL, Pu QZ, Yin JH,et al. Genetic study of a heredity spinocerebellar ataxia pedigree in Hunan province[J]. Chin J Birth Health Hered, 2013, 21(10): 18-20. [舒安利, 蒲泉州, 尹佳慧, 等. 一个遗传性脊髓小脑共济失调大家系的连锁分析[J]. 中国优生与遗传杂志, 2013, 21(10): 18-20.]
Molecular genetics of a Chinese family with spinocerebellar ataxia
WU Dan-dan1, LIU Xiao-xuan2, LIU Jian-yuan3, LIU Yong1, TAO Xiang-ping2, YANG Shu-guang1*, FAN Dong-sheng2*, LIU Shao-jun1*1Department of Neurobiology, Institute of Basic Medical Sciences, Academy of Military Medical Science, Beijing 100850, China
2Department of Neurology,Third Hospital Peking University, Beijing 100191, China
3Department of General Surgery, First People's Hospital of Jiayuguan, Jiayuguan, Gansu 735100, China
*< class="emphasis_italic">Corresponding author. YANG Shu-guang, E-mail: lionamms@hotmail.com; FAN Dong-sheng, E-mail: dsfan@sina.com; LIU Shaojun, E-mail: liusj@bmi.ac.cn
. YANG Shu-guang, E-mail: lionamms@hotmail.com; FAN Dong-sheng, E-mail: dsfan@sina.com; LIU Shaojun, E-mail: liusj@bmi.ac.cn
This work was supported by the National Natural Science Foundation of China (81370051, 81471155)
ObjectiveTo study the genotype of the members of a Chinese family with spinocerebellar ataxia (SCA).MethodsThe peripheral blood samples of 6 patients and 40 asymptomatic people belonged to the family were collected. Referring to the clinical manifestations of the proband and second-generation sequencing results, the CAG trinucleotide repeats of the pathogenic gene ATXN2 were amplified by polymerase chain reaction (PCR). The repeated times of the trinucleotide in normally and abnormally amplified alleles were defined by agarose gel electrophoresis and PCR products sequencing.ResultsAutosomal dominant heredity was the cause of the SCA in this family. Six out of 46 in the fourth-generation were SCA2 patients, 7 were the carriers of pathogenic allele. The repeated times of CAG trinucleotide were within the normal range in one of the two alleles of ATXN2, but they were in abnormal range in the another one. The repeated times of CAG trinucleotide were 40-46 in abnormal alleles of patients.ConclusionAutosomal dominant heredity SCA2 has been diagnosed in this family caused by the dynamic nutation of CAG trinucleotide repeats, and 7 pathogenic allele carriers in this family were confirmed by genetic diagnosis.
spinocerebellar ataxias; trinucleotide repeats; dynamic mutation; genetic diagnosis
R744.7
A
0577-7402(2015)08-0638-05
10.11855/j.issn.0577-7402.2015.08.07
2015-01-20;
2015-06-28)
(责任编辑:沈宁)
国家自然科学基金(81370051,81471155)
吴丹丹,硕士研究生。主要从事家族性神经系统遗传疾病的研究
100850 北京 军事医学科学院基础医学研究所神经生物学实验室(吴丹丹、刘勇、杨树广、刘少君);100191北京 北京大学第三医院神经科(刘小璇、陶襄萍、樊东升);735100 甘肃嘉峪关 嘉峪关市第一人民医院普外科(刘建元)
杨树广,E-mail:lionamms@hotmail.com;樊东升,E-mail:dsfan@sina.com;刘少君,E-mail:liusj@bmi.ac.cn
