以活性炭为载体制备Pd和Pt加氢脱硫催化剂
2015-06-24王安杰陈永英
董 超,李 翔,2,王安杰,2,陈永英
(1.大连理工大学 精细化工国家重点实验室, 辽宁 大连 116012;2. 辽宁省高校石油化工技术与装备重点实验室, 辽宁 大连 116012)
以活性炭为载体制备Pd和Pt加氢脱硫催化剂
董 超1,李 翔1,2,王安杰1,2,陈永英1
(1.大连理工大学 精细化工国家重点实验室, 辽宁 大连 116012;2. 辽宁省高校石油化工技术与装备重点实验室, 辽宁 大连 116012)
分别以椰壳活性炭、果壳活性炭和木质活性炭为载体,采用等体积浸渍法制备了Pd/C和Pt/C催化剂。以二苯并噻吩(DBT)为模型含硫化合物,考察了不同种类活性炭负载的贵金属催化剂加氢脱硫(HDS)催化性能。结果表明,增加活性炭表面酸性含氧基团或碱性基团数量都有助于提高Pt和Pd的分散度。DBT在Pd/C和Pt/C催化剂催化下进行HDS反应时,直接脱硫(DDS)路径选择性高于加氢反应路径(HYD)选择性,其中Pt/C催化剂的HDS催化活性和DDS路径选择性都显著高于Pd/C催化剂。Pd/C和Pt/C催化剂的HDS催化性能主要取决于载体表面官能团的种类和分布。Pd和Pt催化剂的HYD反应路径选择性和稳定性都随载体表面酸性含氧基团的增加而增加,但它们断裂C—S的活性却有所降低;增加载体表面碱性基团数量则有助于提高催化剂断裂C—S的活性,但不利于其稳定性。
钯;铂;活性炭;加氢脱硫
油品的深度脱硫是清洁燃料生产所面临的一个重要课题。在炼油厂中,油品中的硫主要通过加氢精制工艺中的加氢脱硫(HDS)过程脱除,传统的催化剂为负载型Co-Mo或Ni-Mo等双金属硫化物。大分子的二苯并噻吩(DBT)及其烷基取代物是馏分油中最难脱除的含硫化合物。这些芳香杂环含硫化合物分子具有稳定的平面共轭结构,并且由于空间位阻效应,其中的硫原子难以接近层状结构硫化物催化剂的活性中心,因而反应活性很低。当DBT类含硫化合物中的芳环加氢后,分子结构发生扭曲,空间位阻降低,其HDS活性得到显著提高。因此,提高催化剂加氢活性是提高其HDS活性的途径之一。负载型贵金属催化剂如Pd和Pt等催化剂具有良好的低温加氢催化性能,在针对DBT等芳香杂环类含硫化合物的深度加氢脱硫中展现出良好的应用前景[1-2]。载体是贵金属催化剂的重要组成部分,其物理化学性质,尤其是表面酸碱性质是影响贵金属催化剂催化性能的主要因素之一。大量研究表明,提高载体的酸性有利于提高贵金属催化剂HDS活性和耐硫性能[3]。
活性炭是一类重要的贵金属催化剂载体。它具有来源广泛、价格低廉、比表面积大、孔隙结构发达、表面性质可调等特点。活性炭表面丰富的含氧基团能够显著影响贵金属催化剂的性能[4]。表面含氧官能团大致可分为两类,即酸性的羧基、内酯基、羟基、羰基等基团和碱性的吡喃酮类等基团[5]。活性炭表面基团的种类和数量与其原材料、活化条件、预处理方法及使用过程中的氧化还原反应条件密切相关。笔者选取具有不同表面基团分布特征的椰壳活性炭、果壳活性炭和木质活性炭作载体,制备了Pd和Pt催化剂,以DBT为模型含硫化合物,研究了载体表面性质尤其是酸碱性质对贵金属催化剂HDS催化性能的影响。
1 实验部分
1.1 原料
椰壳活性炭,购自海南椰球实业有限公司,记为C-a;果壳活性炭,购自巩义清新活性炭公司和华北地区特种化学试剂开发中心,分别记为C-b和C-c;木质活性炭,购自天津市科密欧化学试剂有限公司,记为C-d。氯化钯(PdCl2)、氯铂酸(H2PtCl6),分析纯,天津化学试剂一厂产品。二苯并噻吩(DBT),由联苯和硫合成[6]。十氢萘,分析纯,购自上海试剂分装厂。
1.2 催化剂前体的制备
采用等体积浸渍法制备催化剂。将载体用去离子水煮沸30 min,干燥。将计量的PdCl2盐酸溶液或H2PtCl6水溶液滴加到载体中,边滴加边搅拌。滴加完后,在室温下浸渍8 h,然后在95℃真空干燥,制得催化剂前体。Pd或Pt的负载量以质量分数计,均为0.5%。
1.3 HDS反应
采用内径8 mm的固定床反应器进行HDS反应。催化剂经压片、破碎至20~40目装填,用量为0.05 g。HDS反应前,在300℃下,用H2以75 mL/min流量对催化剂前体还原1 h。然后在压力5.0 MPa、重时空速MHSV为54 h-1、氢/油体积比1500、300℃的条件下进行HDS反应。反应原料为质量分数0.8%的DBT-十氢萘溶液。采用配有Agilent公司HP-5毛细管色谱柱的HP-6890N型气相色谱仪测定原料和HDS反应产物组成,固定相为5%二苯基-95%二甲基聚硅氧烷。
DBT的HDS反应主要通过直接脱硫(DDS)和加氢(HYD)2条并行的反应路径进行。联苯(BP)是DDS反应路径的唯一产物,因为在有机含硫化合物如DBT存在的情况下,BP很难进一步加氢生成苯基环己烷(CHB)[7]。四氢二苯并噻吩(TH-DBT)和六氢二苯并噻吩(HH-DBT)是HYD反应路径脱硫的主要含硫中间体,再脱硫后分别生成CHB和联环己烷(BCH)。DBT在活性炭负载的Pd和Pt催化剂催化下的反应产物中除脱硫产物外,还能检测到TH-DBT和HH-DBT等含硫中间体。因此,用DBT加氢脱硫转化率(xHDS)描述催化剂的脱硫性能,由式(1)计算。
xHDS=(CDBT,0-CDBT-CTH-CHH)/CDBT,0×100%
(1)
式(1)中,CDBT,0和CDBT分别为反应器入口和出口处DBT浓度,mmol/L;CTH和CHH分别为反应器出口处TH-DBT和HH-DBT的浓度,mmol/L。催化剂DDS路径选择性可用BP的选择性(sBP)表示,催化剂HYD路径的选择性用(1-sBP)表示。
1.4 载体和催化剂的表征
采用Boehm滴定法测定活性炭表面含氧基团。分别称取0.5 g活性炭加到25 mL浓度为0.05 mol/L的C2H5ONa溶液、NaOH溶液、Na2CO3溶液或NaHCO3溶液中,常温搅拌2 h后放置2 d。分别抽取15 mL上层清液,加入25 mL浓度为0.05 mol/L的稀盐酸,然后用0.05 mol/L的NaOH溶液返滴定。NaHCO3可中和活性炭表面的羧基类基团,Na2CO3可中和羧基和内酯基类基团,NaOH可中和羧基、内酯基和羟基类基团,而C2H5ONa碱性最强,可中和活性炭表面所有的酸性含氧基团[8-9]。测定活性炭表面碱性时,将0.5g活性炭样品加到25 mL浓度为0.05 mol/L的盐酸溶液中,常温搅拌2 h后放置2 d。抽取15 mL上层清液,加入25 mL浓度为0.05 mol/L的NaOH溶液,然后用0.05 mol/L的盐酸溶液返滴定[10]。
采用美国Micrometrics TristarⅡ3020吸附仪测定催化剂N2吸附-脱附等温线,测定前样品在300℃下脱气3 h。采用Chembet-3000型分析仪动态脉冲法测定H2化学吸附[11]。将1 g样品装填到U型反应管中,首先在室温下(25℃)用50 mL/min的高纯Ar吹扫30 min,然后在50 mL/min的纯H2气氛下以10℃/min的速率升温至300℃,停留1 h。然后在相同温度下,用50 mL/min的高纯Ar吹扫1 h,脱除物理吸附的H2,然后降至70℃进行测定。将载气高纯Ar流量固定在50 mL/min,向反应管内反复脉冲注入100 μL的H2-Ar混合气(H2体积分数10%),直至吸附饱和。计算H2吸附量、活性组分分散度以及贵金属平均粒径[11]。
2 结果与讨论
2.1 活性炭负载Pt、Pd催化剂及其载体的表征结果
表1列出了不同活性炭样品Boehm滴定和N2物理吸附结果。由表1可见,椰壳活性炭C-a比表面积最大,两种果壳活性炭C-c、C-b比表面积相当,都小于木质活性炭C-d和椰壳活性炭C-a;在4种活性炭样品中,果壳活性炭C-b的表面酸性含氧基团最为丰富,其次为椰壳活性炭C-a,能检测到除内酯基外其它酸性含氧基团,木质活性炭C-d和果壳活性炭C-c表面酸性含氧基团分布类似,主要为羰基,C-c表面还有少量的羟基。这些活性炭载体表面总酸量按C-b、C-a、C-c、C-d顺序降低,而总的碱量则按C-d、C-b、C-a、C-c顺序降低。木质活性炭C-d表面总酸量最低,但总碱量最高,并且总碱量大于总酸量。果壳活性炭C-c表面主要为酸性的含氧官能团,只检测到少量的碱性基团。

表1 不同活性炭载体的比表面积(SBET)及表面含氧基团的量(ρO-group)
根据H2化学吸附实验结果计算了不同活性炭载体负载的Pd和Pt催化剂活性组分粒径和分散度,结果列于表2。由表2可以看出,这些活性炭负载的Pd和Pt催化剂氢化学吸附量变化规律一致:果壳活性炭C-b负载的Pd和Pt的氢吸附量最大,相应的分散度最大,粒子尺寸最小。而以果壳活性炭C-c负载的Pd和Pt催化剂氢吸附量以及活性组分分散度最小,相应的粒子尺寸最大。椰壳活性炭C-a和木质活性炭C-d负载的催化剂分散度相当。结合表1可以看出,金属活性组分的分散度与载体比表面积之间没有直接关系。C-b比表面积最小,但其负载的Pd和Pt催化剂分散度却最高。活性炭表面酸性含氧基团是影响贵金属活性组分分散度的一个重要因素。一般认为表面的含氧基团能够降低活性炭的疏水性,改善其亲水性,从而促进浸渍过程中贵金属盐前体的分散[12]。此外,表面含氧基团还可以作为高分散度金属微晶的成核中心[13]。在4种活性炭样品中,C-b表面酸性含氧基团最为丰富、酸量最高(表1),因此贵金属活性组分的分散度最好。另外,活性炭载体表面碱性基团是Pt等金属组分强吸附的锚定位[5]。所以尽管C-d表面酸性含氧基团较少,但碱性基团含量最高(见表1),活性组分的分散度也较高(见表2)。C-c表面总酸量和总碱量都比较低,其活性组分分散度最差。
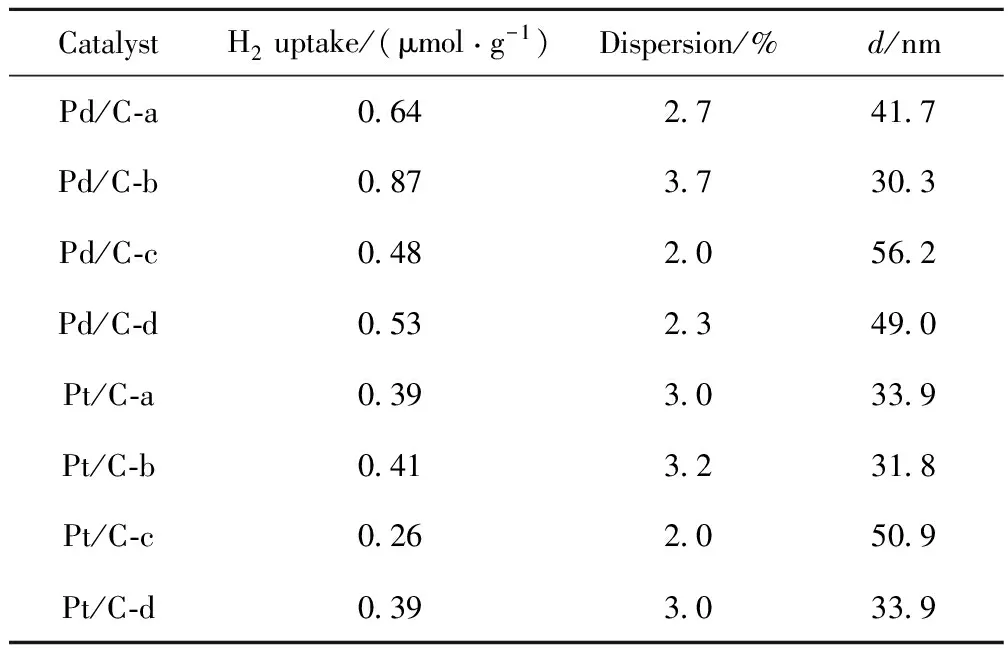
表2 负载型Pd催化剂和Pt催化剂的H2化学吸附结果
2.2 活性炭负载Pt、Pd催化剂的HDS催化活性
图1示出了DBT在不同活性炭负载的Pd催化剂催化下的HDS反应的转化率和反应路径选择性随反应时间的变化,所得产物收率(以其相对分压计)随反应时间的变化示于图2。从图1可见,在这4种催化剂中,Pd/C-d的初始活性最高,DBT的初始转化率(xDBT)以及加氢脱硫转化率(xHDS)分别为52%和44%。Pd/C-b初始活性最低,DBT的初始转化率以及加氢脱硫转化率分别为31%和20%。在Pd/C-a、Pd/C-c和Pd/C-d催化下,DBT主要通过DDS路径脱硫,其中Pd/C-d催化剂的DDS选择性最高,约70%左右。在Pd/C-b催化下,DBT的DDS路径选择性为53%,略高于HYD路径选择性(47%),说明2条反应路径并重。与此相对应,从图2可见,作为DDS路径的唯一产物,BP在Pd/C-d催化剂上收率最高,而在Pd/C-b催化剂上最低。虽然Pd/C-b催化剂初始反应活性较低,但稳定性最好,在反应8 h内DBT转化率、反应路径选择性和各产物收率没有明显变化;Pd/C-c和Pd/C-d稳定性较差,以Pd/C-d为例,反应8 h后DBT转化率和加氢脱硫转化率分别由初始的52%和44%降至37%和27%。
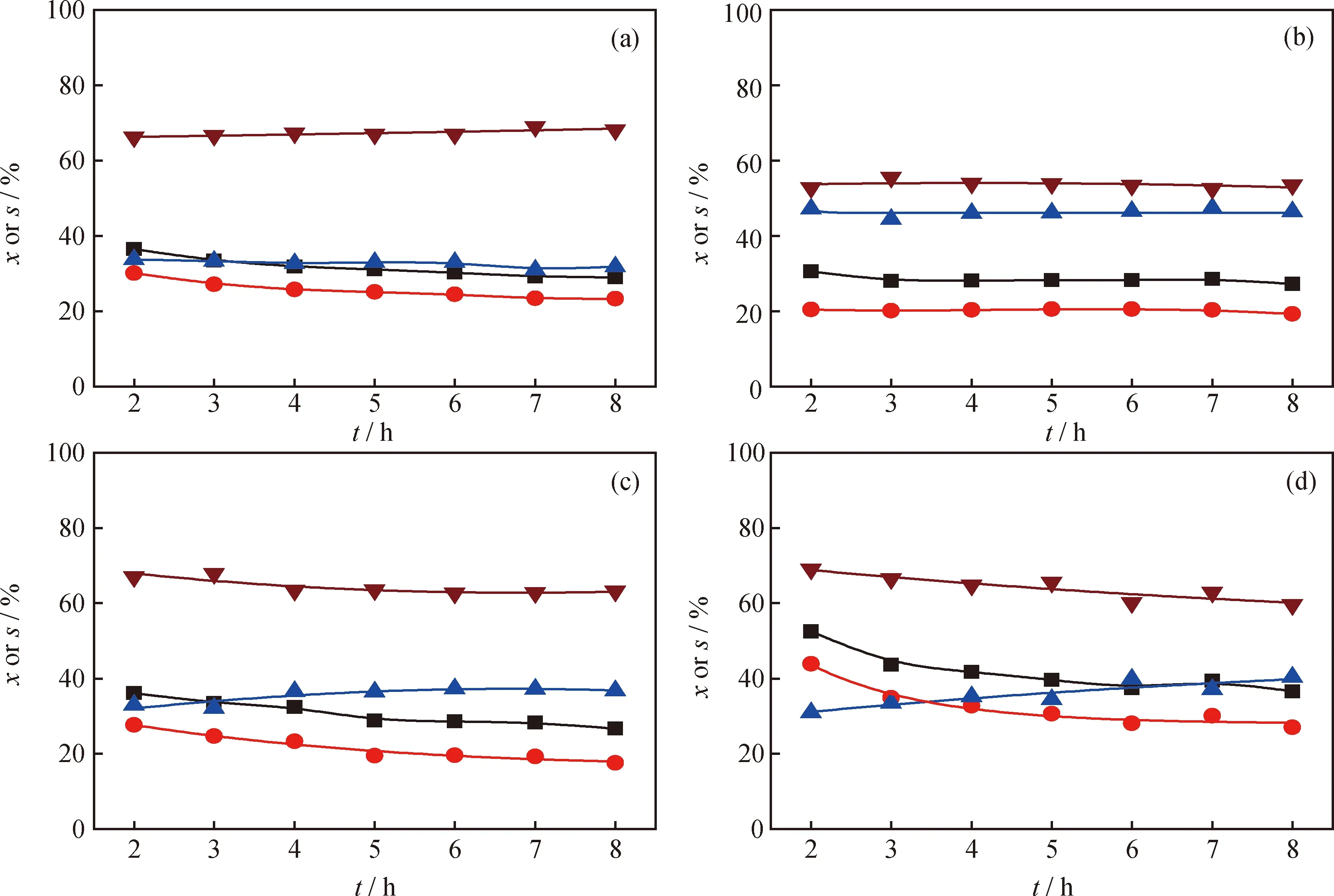
图1 300℃、5 MPa条件下不同活性炭负载的Pd催化剂HDS反应性能
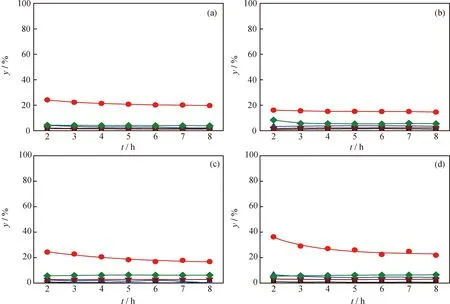
图2 300℃、5 MPa条件下DBT在不同活性炭负载Pd催化剂催化HDS反应产物收率(y)随时间的变化
图3示出了DBT在不同活性炭负载的Pt催化剂催化下的HDS反应的转化率和反应路径选择性随反应时间的变化,所得产物收率(以其相对分压计)随反应时间的变化示于图4。与活性炭负载的Pd催化剂类似,Pt/C-d初始反应活性最高但稳定性最差,Pt/C-b初始活性最低但稳定性较好。Pt/C催化剂对DBT的加氢脱硫活性显著高于Pd/C催化剂。另外,DBT在Pt/C催化剂催化下进行HDS反应时,BP选择性达到90%左右,说明DDS路径占主导地位。由图4可以看出,Pd/C-d催化剂催化所得BP初始收率最高,Pd/C-b催化所得的最低。
在已考察过的贵金属活性组分中,Pd和Pt在催化DBT类含硫化合物HDS反应中表现出很高的活性和最佳的耐硫性能;且二者各有特点,Pt有较高的DDS活性,而Pd加氢活性较高,但脱硫活性比Pt催化剂低[1-2]。与此结论一致,Pt/C催化剂催化所得BP的收率和选择性显著高于Pd/C催化剂,说明Pt/C催化剂直接断裂C—S键的活性明显高于Pd/C催化剂,这可能是造成DBT在2种催化剂上转化率和加氢脱硫转化率差别的主要原因。活性炭载体对Pt和Pd的HDS性能表现出了相同的影响规律。以木质活性炭C-d作载体的催化剂的初始活性最高,但稳定性较差;果壳活性炭C-b负载的催化剂的初始活性较低,但稳定性较佳。这与活性组分分散度之间没有必然的关系(见表2),可能的原因是这些催化剂的金属粒子尺寸较大,表现出类似体相金属的性质,催化性能对金属颗粒尺寸的变化不敏感。结合Boehm滴定结果(见表1)可以看出,催化剂性能与载体表面活性基团的分布有一定联系。总碱量最高的木质活性炭C-d负载的Pt和Pd催化剂的初始HDS活性和DDS活性(BP收率)最高,总酸量最高的果壳活性炭C-b作载体的催化剂的初始HDS活性和DDS活性最低。C-a和C-c负载的催化剂的性能则介于二者之间。
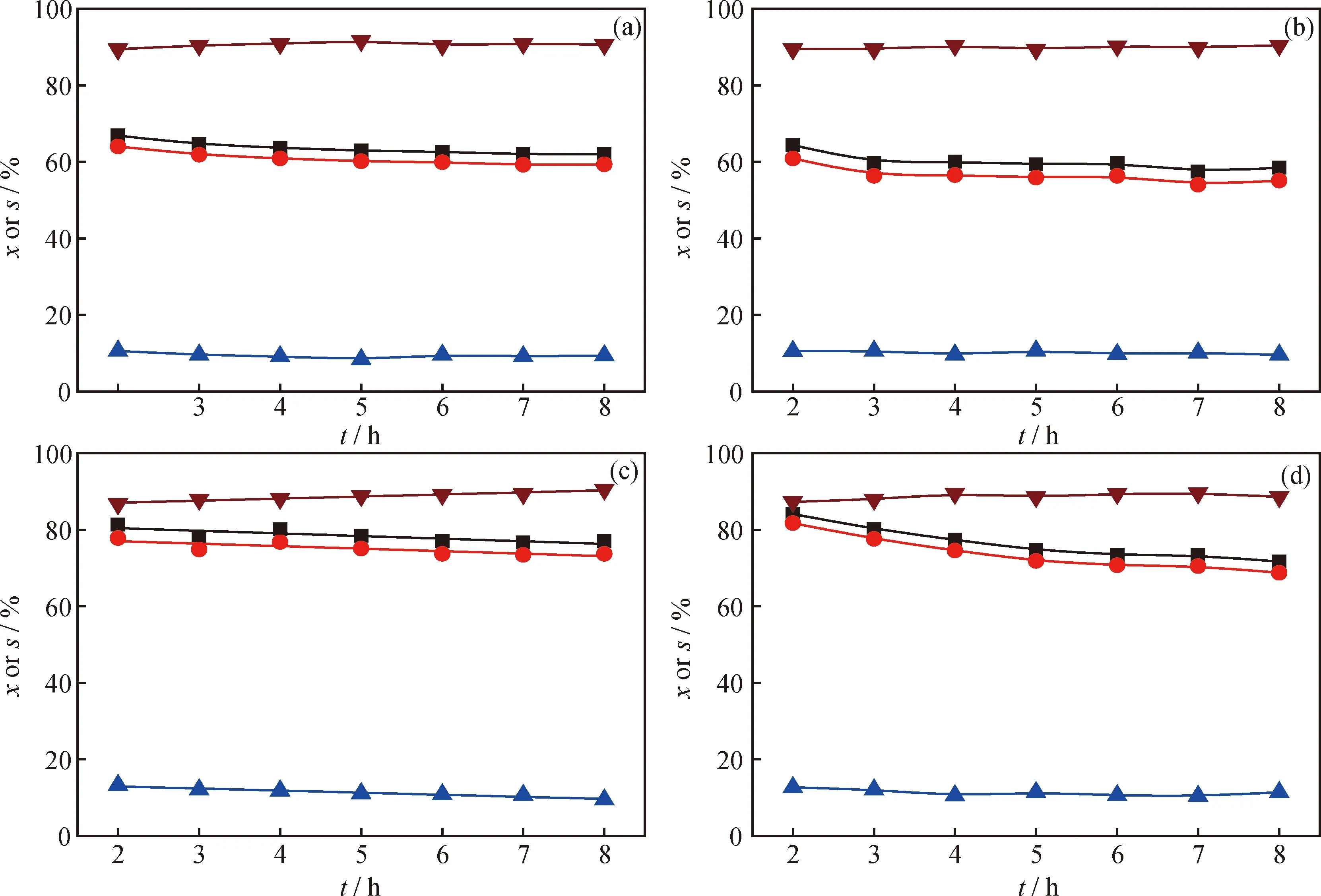
图3 300℃、5 MPa条件下不同活性炭负载的Pt催化剂HDS反应性能

图4 300℃、5 MPa条件下DBT在不同活性炭负载Pt催化剂催化下HDS反应产物收率(y)随时间的变化
目前,有关芳香杂环含硫化合物中C—S键断裂机理尚存争议。氢解[14]和β-消除[15]是两个被广泛采用的机理。氢解机理认为,在C—S断裂的同时形成C—H键和S—H键。噻吩类含硫化合物在金属催化剂催化下的氢解过程可分为两步。首先是金属活性中心通过氧化加成插入到噻吩环的C—S键中,形成1个六元环的硫杂金属配合物,然后再与氢或金属氢化物反应脱硫[16]。β-消除机理则认为,C—S的断裂经历消除反应历程,包括α-C—S键和β-C—H键的断裂,生成H2S和相应的C=C双键[15]。笔者最近针对DBT在Ni2P催化下的反应机理和动力学的研究表明,DBT的HDS反应网络中可能同时包含了氢解和β-消除两种反应机理,DBT和TH-DBT主要通过氢解机理脱硫,而HH-DBT则主要通过β-消除机理断裂C—S键[17]。β-消除过程同时需要碱中心(脱除β-H)以及酸中心或空穴位(吸附反应物分子中的硫)的参与[15]。另外,载体的碱性有助于增加表面上金属颗粒的电子密度[18],这将有助于氧化加成反应的进行。无论从β-消除机理还是氢解机理角度来看,提高载体碱性都有利于C—S键的断裂,这可能是以C-d作载体的催化剂表现出较高脱硫活性的原因。
增加载体酸性是一种常用的提高贵金属催化剂HDS活性、加氢路径选择性以及耐硫性能的手段。载体酸中心与贵金属活性组分之间可能存在电子相互作用,活性中心部分电荷转移到载体酸中心上,导致活性组分电子密度降低,形成所谓的“缺电子结构”(Electron-deficient)[19]。这部分“缺电子”物种具有较强的吸附芳环能力和加氢活性[20],从而提高了催化剂对DBT中芳环的加氢活性。另一方面,S在贵金属上吸附时,金属d轨道电子会转移到电子受体S原子上;降低贵金属活性组分电子云密度会削弱活性组分与S之间的相互作用,提高催化剂耐硫性能[21]。但也有的观点认为,只有粒径小于1.5 nm的贵金属颗粒才有可能形成缺电子结构[22]。这是由于如果活性组分粒度较大,金属颗粒表面电子密度的变化会被颗粒内部大量的原子“稀释”[23]。在本研究中,活性组分的平均颗粒尺寸大于15 nm(见表2),因此,总酸量较高的活性炭如果壳活性炭C-b负载的催化剂较高的HYD路径选择性和较好的稳定性可能不能归因于酸中心和活性组分间的电子相互作用。除形成缺电子结构之外,还可用“氢溢流”理论解释载体酸性对贵金属催化剂性能的调变。氢溢流对多相催化体系有重要影响,如产生新活性中心、抑制催化剂表面的积炭、提高活性组分还原能力、直接参与反应等[24]。提高载体的酸性有利于溢流氢的生成和稳定[25]。比如,Zhang等[26]合成了一种包覆型MCM-41/Y介微孔分子筛,并以其作载体制备了负载型Pt-Pd催化剂,发现该催化剂在萘加氢过程中表现出较好的耐硫性能。他们认为,在微孔孔道内贵金属活性中心表面生成的高活性活化氢物种可以溢流到介孔孔道内,不仅能够促进芳烃的加氢,还能够通过“自清洁”作用,还原被硫化的金属组分,提高催化剂耐硫性能。本研究所得数据尚不足以就活性炭表面酸碱基团对贵金属活性组分催化性能的影响机制作出明确的解释,但由实验数据可以看出,单纯调变载体表面酸性或碱性很难达到全面改善催化剂催化HDS反应性能的目的。综合性能优良的加氢脱硫催化剂载体可能应有均衡的表面酸碱中心分布。
3 结 论
(1) 活性炭载体比表面积不是决定Pd和Pt活性组分分散度的关键因素,但增加活性炭表面酸性含氧基团或碱性基团数量,都有助于提高Pt和Pd的分散度。
(2) DBT在活性炭负载的Pt和Pd催化剂催化下进行HDS反应时,DDS路径选择性高于HYD反应路径,其中Pt催化剂的HDS活性和DDS路径选择性都显著高于Pd催化剂。
(3) 活性炭负载的Pd和Pt催化剂的HDS催化性能主要取决于载体表面含氧官能团的种类和分布,与活性组分分散度没有直接关系。Pd和Pt催化剂的HYD反应路径选择性和稳定性都随载体表面酸性含氧基团的增加而增加,但它们断裂C—S的活性却有所降低;相反,增加载体表面碱性基团数量有助于提高催化剂断裂C—S的活性,但不利于其稳定性。
[1] NIQUILLE-RÖTHLISBERGER A, PRINS R. Hydrodesulfurization of 4,6-dimethyldibenzothiophene and dibenzothiophene over alumina-supported Pt, Pd, and Pt-Pd catalysts[J]. Journal of Catalysis, 2006, 242(1): 207-216.
[2] NIQUILLE-RÖTHLISBERGER A, PRINS R. Intermediates in the hydrodesulfurization of 4,6-dimethyl-dibenzothiophene over Pd/γ-Al2O3[J]. Journal of Catalysis, 2005, 235(1): 229-240.
[3] SUN Y Y and PRINS R. Hydrodesulfurization of 4,6-dimethyldibenzothiophene over noble metals supported on mesoporous zeolites[J]. Angewandte Chemie International Edition, 2008, 47(44): 8478-8481.
[4] 李晓芸, 马丁, 包信和. 不同活性炭上Pt催化剂的分散性及其在甲基环己烷脱氢反应中的催化性能[J]. 催化学报, 2008, 29(3): 259-263. (LI Xiaoyun, MA Ding, BAO Xinhe. Dispersion of Pt catalysts supported on activated carbon and their catalytic performance in methylcyclohexane dehydrogenation[J]. Chinese Journal of Catalysis, 2008, 29(3): 259-263. )
[5] FRAGA M A, JORDO E, MENDES M J, et al. Properties of carbon-supported platinum catalysts: Role of carbon surface sites[J]. Journal of Catalysis, 2002, 209(2): 355-364.
[6] QIAN W H, ISHIHARA A, OGAWA S, et al. Study of hydrodesulfurization by the use of35S-labeled dibenzothiophene. 1. Hydrodesulfurization mechanism on sulfided Mo/Al2O3[J]. The Journal of Physical Chemistry, 1994, 98(3): 907-911.
[7] REN J, WANG A J, LI X, et al. Hydrodesulfurization of dibenzothiophene catalyzed by Ni-Mo sulfides supported on a mixture of MCM-41 and HY zeolite[J]. Applied Catalysis A: General, 2008, 344(1-2): 175-182.
[8] LI Y H, WANG S G, LUAN Z K, et al. Adsorption of cadmium(II) from aqueous solution by surface oxidized carbon nanotubes[J]. Carbon, 2003, 41(5): 1057-1062.
[9] LU X C, JIANG J C, SUN K, et al. Surface modification, characterization and adsorptive properties of a coconut activated carbon[J]. Applied Surface Science, 2012, 258(20): 8247-8252.
[10] LU C, SU F S, HU S. Surface modification of carbon nanotubes for enhancing BTEX adsorption from aqueous solutions[J]. Applied Surface Science, 2008, 254(21): 7035-7041.
[13] AUER E, FREUND A, PIETSCH J, et al. Carbons as supports for industrial precious metal catalysts[J]. Applied Catalysis A: General, 1998, 173(2): 259-271.
[14] TODOROVA T, PRINS R, WEBER Th. A density functional theory study of the hydrogenolysis reaction of CH3SH to CH4on the catalytically active(100)edge of 2H-MoS2[J].Journal of Catalysis,2005, 236(2):190-204.
[15] BATAILLE F, LEMBERTON J, MICHAUD P, et al. Alkyldibenzothiophenes hydrodesulfurization-promoter effect, reactivity, and reaction mechanism[J]. Journal of Catalysis, 2000, 191(2): 409-422.
[16] GARCIA J J, AREVALO A, CAPELLA A, et al. Analysis of a hydrodesulfurization process- 2[1]. The reactions of 2- and 3-methylthiophenes with tris(triethylphosphine)platinum(0)[J]. Polyhedron, 1997, 16(18): 3185-3195.
[17] LI X, BAI J, WANG A, et al. Hydrodesulfurization of dibenzothiophene and its hydrogenated intermediates over bulk Ni2P[J]. Topics in Catalysis, 2011, 54(5-7): 290-298.
[18] SHI C K, YANG L F, WANG Z C, et al. Promotion effects of ZrO2on the Pd/HZSM-5 catalyst for low-temperature catalytic combustion of methane[J]. Applied Catalysis A: General, 2003, 243(2): 379-388.
[19] SHEU L L, KNÖZINGER H, SACHTLER W M H. Palladium carbonyl clusters entrapped in NaY zeolite cages: Ligand dissociation and cluster-wall interactions[J]. Journal of the American Chemical Society, 1989, 111(21): 8125-8131.
[20] YOSHIMURA Y, TOBA M, MATSUI T, et al. Active phases and sulfur tolerance of bimetallic Pd-Pt catalysts used for hydrotreatment[J]. Applied Catalysis A: General, 2007, 322: 152-171.
[21] YASUDA H, MATSUBAYASHI N, SATO T, et al. Confirmation of sulfur tolerance of bimetallic Pd-Pt supported on highly acidic USY zeolite by EXAFS[J]. Catalysis Letters, 1998, 54(1-2): 23-27.
[22] REYES P, OPORTUS M, PECCHI G, et al. Influence of the nature of the platinum on the surface properties and catalytic activity of alumina-supported catalysts[J]. Catalysis Letters, 1996, 37(3-4): 193-197.
[23] STAKHEEV A Y, KUSTOV L M. Effects of the support on the morphology and electronic properties of supported metal clusters: modern concepts and progress in 1990s[J]. Applied Catalysis A: General, 1999, 188(1-2): 3-35.
[24] CONNER W C, FALCONER J L. Spillover in heterogeneous catalysis[J]. Chemical Reviews, 1995, 95(3): 759-788.
[25] MILLER J T, MEYERS B L, MODICA F S, et al. Hydrogen temperature-programmed desorption (H2TPD) of supported platinum catalysts[J]. Journal of Catalysis, 1993, 143(2): 395-408.
[26] ZHANG H J, MENG X C, LI Y D, et al. MCM-41 overgrown on Y composite zeolite as support of Pd-Pt catalyst for hydrogenation of polyaromatic compounds[J]. Industrial & Engineering Chemistry Research, 2007, 46(12): 4186-4192.
Preparation of Pd and Pt Hydrodesulfurization Catalysts Supported on Activated Carbons
DONG Chao1, LI Xiang1, 2, WANG Anjie1, 2, CHEN Yongying1
(1.StateKeyLaboratoryofFineChemical,DalianUniversityofTechnology,Dalian116012,China;2.LiaoningKeyLaboratoryofPetrochemicalTechnologyandExperiment,Dalian116012,China)
The Pd and Pt hydrodesulfurization (HDS) catalysts supported on coconut shell activated carbon, nut shell activated carbon, and wood activated carbon were prepared by an incipient wetness impregnation method. The HDS catalytic performances of these noble metal catalysts supported on different activated carbons were investigated by using a model feed containing 0.8 % dibenzothiophene (DBT) in decalin. The results indicated that the dispersion of Pd and Pt could be enhanced by increasing the amounts of either the acidic oxygen-containing groups or the basic groups on the surface of the support. In the HDS of DBT over the Pd/C and Pt/C catalysts, the selectivity toward the direct desulfurization pathway (DDS) was higher than that toward the hydrogenation pathway (HYD). Both the HDS activity and the DDS pathway selectivity of the supported Pt catalysts were significantly higher than those of the supported Pd catalysts. The HDS catalytic performances of the Pt/C and Pd/C catalysts were mainly determined by the surface groups of the support and their distributions. An increase of the surface acidic oxygen-containing groups led to an increase of the HYD selectivity and the stability of the Pd and Pt catalysts, but a decrease in their C-S bond cleavage activity. On the other hand, the basic groups showed a positive effect on the C-S bond cleavage activity of Pd/C and Pt/C and a negative influence on their stability.
palladium; platinum; activated carbon; hydrodesulfurization
2014-10-08
国家自然科学基金项目(20973030,21073022,和U1162203)、中央高校基本科研业务费专项资金(DUT13LK18)、辽宁省教育厅科学研究项目(L2013023)及中国石油科技创新项目(2014D-5006-0402)资助 第一作者: 董超,男,博士研究生,从事贵金属催化剂加氢脱硫及脱氮研究
李翔,男,副教授,工学博士,从事加氢精制催化剂及介孔分子筛研究,Tel:0411-84986124;E-mail:lixiang@dlut.edu.cn
1001-8719(2015)02-0542-08
TE624.9
A
10.3969/j.issn.1001-8719.2015.02.035
