丙烷脱氢制丙烯催化剂研究的进展
2015-06-24张凌峰刘亚录胡忠攀杨玉旺于海斌袁忠勇
张凌峰,刘亚录,胡忠攀,杨玉旺,于海斌,袁忠勇
(1.南开大学 化学学院 先进能源材料化学教育部重点实验室 天津化学化工协同创新中心, 天津 300071;2.中海油 天津化工研究设计院, 天津 300131)
丙烷脱氢制丙烯催化剂研究的进展
张凌峰1,刘亚录1,胡忠攀1,杨玉旺2,于海斌2,袁忠勇1
(1.南开大学 化学学院 先进能源材料化学教育部重点实验室 天津化学化工协同创新中心, 天津 300071;2.中海油 天津化工研究设计院, 天津 300131)
丙烯是化工行业重要的基础原料,受丙烯下游产品的拉动,国内外对丙烯的需求量逐年递增。丙烯主要来源于石油的催化裂解,但石油资源的日益匮乏无法满足全球对丙烯的需求,因此研究丙烷脱氢制丙烯工艺具有重大的实际意义。笔者综述了丙烷通过直接脱氢、氧气或二氧化碳氧化脱氢等方法制丙烯的反应热力学、反应机理及常用催化剂体系,论述了各种催化剂的作用机制、催化剂表面的活性物种和性质,评价了它们的丙烷脱氢反应性能,并展望了丙烷脱氢制丙烯的发展方向。
丙烷;丙烯;脱氢;催化剂
丙烯是仅次于乙烯的重要有机石油化工基础原料,广泛用于生产聚丙烯、丙烯醛、丙烯酸、甘油、异丙醇、聚丙烯腈、丁辛醇等化工产品。近年来,随着市场经济的发展,丙烯下游产品的需求量迅速上涨,极大地促进了全球对丙烯的需求。
目前,丙烯绝大部分来源于石油的催化裂化和柴油、石脑油的裂解。裂解法受到丙烷-乙烯联产比例的限制,催化裂化法则受到轻质烃进一步制取高辛烷值汽油的制约[1];而且,随着石油资源的日渐匮乏,传统的丙烯生产技术已无法满足日益增长的丙烯需求,寻求新的丙烯生产技术已成为石油化工行业的主要发展趋势。丙烷脱氢制丙烯成为增加丙烯来源的重要途径之一。石油和天然气资源中含有大量的丙烷,如油田中丙烷约占6%,液化石油气中约占60%,湿天然气中可达15%[2],炼厂气中也含有一定量的丙烷等。但目前丙烷的主要用途是作为燃料,或者在尾气中被直接放空,造成很大的资源浪费;将丙烷转化为丙烯,然后制成其他化工产品将会极大地提高丙烷的利用效率。
丙烷脱氢技术主要分为直接脱氢和氧化脱氢,其中直接脱氢技术已经于20世纪90年代实现了工业化生产[3],表1列出了国内已建和拟建的丙烷脱氢项目。与其它丙烯生产技术相比,直接脱氢技术具有总收率高、设备费用较低等优点。但由于丙烷脱氢为强吸热反应(C3H8→C3H6+H2,ΔH=124 kJ/mol),故需在700℃左右的高温下才能有效地进行[4],而高温将导致丙烷的深度裂解和深度脱氢,使丙烯的选择性降低,同时催化剂易因结焦而失活。丙烷氧化脱氢则是以较低温度下的放热反应替代直接脱氢的高温吸热反应,从而大大降低反应的能耗,丙烷氧气氧化脱氢反应C3H8+1/2O2→C3H6+H2O的ΔH=-116.8 kJ/mol。由于不受热力学平衡的限制,反应过程中催化剂不易积炭,避免了催化剂的反复再生,降低了设备投资。然而,反应物C3H8的C—H键能为401.3 kJ/mol,而产物C3H6的C—H键能为360.7 kJ/mol,产物C3H6不易从催化剂表面脱附[5],容易导致丙烯继续深度氧化为CO2、CO等,使丙烯选择性降低。因此开发高活性、高选择性的催化剂是该技术的关键,成为近几年的热点研究课题。笔者综述了几种常见的丙烷脱氢制丙烯反应,包括直接脱氢、氧气氧化脱氢和二氧化碳氧化脱氢,简要介绍了各类反应的热力学和反应机理,并分类讨论了反应常用的催化剂,比较其优、缺点,展望其发展方向。

表1 国内已建和拟建丙烷脱氢项目
1 丙烷直接脱氢制丙烯
丙烷直接脱氢制丙烯反应为热裂解反应,是吸热过程[5],热力学上不易进行,500℃时平衡转化率为18%,600℃时为50%左右。为了获得较高的丙烷转化率,反应温度一般需控制在700℃以上。但是,高温操作不易控制,因为在较高温度下C—C键比C—H键更容易断裂,因此丙烷容易裂解生成甲烷、乙烷和乙烯,且生成的丙烯还会进一步脱氢聚合成多环芳烃等深度脱氢产物,不仅丙烯产率低,还造成催化剂结焦失活。另外,增加反应体系压力也不利于丙烷脱氢反应[6],如在反应温度590℃,101325Pa时,丙烷的转化率达到43%,此时产物中丙烯的摩尔分数只占30%,而在0.01 MPa下,产物中丙烯的摩尔分数可以提高到45%。
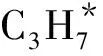
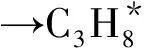
(1)
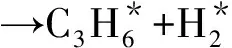
(2)
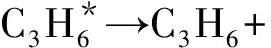
(3)

(4)
Gascón等[8]以Cr2O3/Al2O3为催化剂,采用内径为8 mm的石英管反应器进行催化丙烷脱氢制丙烯反应。他们根据丙烷脱氢氧化反应的特点,利用Langmuir-Hinshelood原理,首先假定催化剂表面都是均匀的、理想的,所有被吸附的丙烷分子在催化剂表面的吸附平衡都满足Langmuir吸附等温式;吸附在催化剂表面的丙烷分子形成表面络合物,而这些表面络合物之间通过相互作用进一步反应,转化为吸附态的丙烯分子,最后吸附态的丙烯分子从理想的催化剂表面脱附,得到气相的丙烯,反应历程如图1所示。
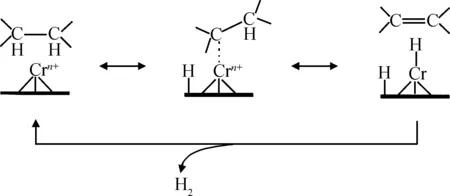
图1 Cr2O3/Al2O3催化剂丙烷脱氢反应机理
丙烷直接脱氢制丙烯反应主要的催化剂有铬基催化剂、铂基催化剂以及新兴的非金属碳催化剂,表2为常见的丙烷直接脱氢制丙烯催化剂的制备方法及其催化性能。

表2 丙烷直接脱氢制丙烯催化剂的催化性能
1.1 Cr系催化剂
丙烷催化脱氢的Catofin、Linde、FBD工艺使用的都是Cr系催化剂,且其一般负载在稳定性较高的Al2O3上。该脱氢过程通常在高于550℃进行,压力为(3~5)×104Pa,单程转化率在48%~65%,反应周期在15~30 min区间。然而,高温会使催化剂迅速失活,因为大量的积炭覆盖了催化剂的活性位。因此,每隔7~15 min需对催化剂进行氧化再生。对于Cr2O3/Al2O3催化剂的研究主要集中在丙烷脱氢的活性位点,因为Cr在Al2O3表面以多种价态(Cr6+,Cr5+,Cr3+和Cr2+)及相态存在[19-20],其中的Cr6+物种很少被作为活性位进行讨论。为了研究氧化态和还原态的Cr对催化活性的影响,Sokolov[21]等对氧化态的CrOx/Al2O3催化剂用H2预还原处理后,直接催化丙烷脱氢。在反应的初始阶段,氧化态的催化剂体系中检测到少量的CO,而还原态的催化剂体系中则没有发现该物质,说明氧化态的Cr基催化剂中的晶格氧参与了反应,并发生了深度氧化脱氢。另外,氧化态催化剂的催化活性比还原态的高,表明Cr6+具有更高的催化性能。随着反应的进行,晶格氧被消耗,氧化态催化剂表现出较快的失活行为,最终2种催化剂的催化活性相当。原位紫外漫反射分析表明,在反应过程中,Cr6+逐渐被丙烷还原为Cr3+,成为最终的活性中心。
虽然许多研究者将Cr系催化剂脱氢活性中心归咎于配位不饱和的Cr3+[22-30],但是,究竟何种类型的Cr3+是脱氢活性中心,至今仍没有一致结论,这也是值得继续关注和解决的问题。根据不同的来源,Cr3+物种可分为“还原Cr3+”和“非还原Cr3+”。Puurunen等[22]认为,非还原Cr3+和Cr3+簇的催化活性比还原Cr3+和孤立的Cr3+要高;而Rossi和Hakuli等[26-27]提出,单核还原Cr3+是活性中心;Hakuli等[25,27]则认为,在催化剂表面负载量小于5 atom Cr/nm2时,还原Cr3+是活性中心,而当负载量超过5 atom Cr/nm2时,非还原Cr也是脱氢的还原中心。Cr3+、Cr5+和Cr6+可以形成多种化合物,并具有不同的还原性和催化行为[29]。Cr5+物种和催化剂的初活性相关,但主活性中心是Cr2+,而丙烯的选择性主要由Cr3+物种决定[30]。也有研究者认为,在烃类脱氢反应中,催化剂表面活性中心是Cr2+而不是Cr3+。
在不同的载体上,Cr物种的催化活性也存在着差异。Kumar等[10]通过对催化剂进行一系列的光谱表征得出,在以SBA-15为载体的Cr催化剂中,大于四配位的孤立的Cr(Ⅲ)比表面晶体Cr2O3中的Cr具有更高的活性、选择性和稳定性;而以Al2O3为载体的Cr催化剂中,聚合的Cr物种则比孤立Cr活性更强。由于每一种载体都具有独特的物理化学性质,比如机械强度、孔道结构以及酸碱性等,活性物种与不同载体之间的相互作用力不一样,催化剂在不同的载体上表现出不同的分散性且存在形式也不一样,直接影响到催化剂的活性。所以,选择合适的载体,并针对不同载体的物化性质设计最佳的催化剂制备方案,对进一步改善催化剂的催化活性至关重要。
催化剂积炭是影响该催化剂体系活性的核心问题。在丙烷直接脱氢制丙烯工艺中,需要对使用的催化剂频繁地氧化再生。催化剂经历多次再生之后,催化活性不能到达其原始水平,因为活性物种的结构发生了变化,产生不可逆失活[21]。显然,如何减少催化剂积炭也是该催化反应当前面临的一个主要问题,比如将积炭迅速转移到载体上而不影响活性位点发挥作用,从而延长催化剂再生的周期。
Cr系催化剂对原料中杂质的要求比较低,反应活性高且价格低廉,原料易得,与贵金属催化剂相比具有很大优势。但是,Cr系催化剂中的Cr是重金属组分,容易污染环境,而且在反应中催化剂失活较快需要反复再生,工业操作相对麻烦,因此,开发抗失活的Cr系催化剂是目前丙烷脱氢行业面临的巨大挑战和亟待解决的问题。
1.2 Pt系催化剂
对于Pt系催化剂,催化剂粒径是影响其对丙烷直接脱氢制丙烯催化反应性能的一个重要因素[31]。催化反应过程中,小颗粒催化剂有利于C—C键断裂,易于发生裂解反应,从而使反应活性非常高,但积炭现象非常严重,丙烯的选择性低;大颗粒催化剂则对C—H键断裂反应的选择性较好,使得催化剂具有高的丙烯选择性和较低的积炭量,但活性不如小颗粒催化剂。所以,需要调控催化剂颗粒大小,合成颗粒大小合适的催化剂,以同时提高其反应活性和对丙烯的选择性,同时设法减少积炭量。
同样,载体也对Pt系催化剂催化丙烷脱氢反应产生重要的影响。不同的载体具有不同的表面性质,Pt与载体之间相互作用也存在着差异。Zhang等[32]讨论了ZSM-5、γ-Al2O3、介孔氧化铝和SBA-15 4种载体对其负载的Pt催化剂催化脱氢活性的影响。Pt在介孔结构氧化铝的载体上可以形成较为均一的粒子分布,在催化脱氢反应中呈现出最佳的催化效果;ZSM-5具有较强的酸性位,容易引发裂解反应,导致丙烯的选择性非常低;多孔结构的γ-Al2O3载体则使得积炭更容易聚集,覆盖了活性位点,使得催化活性下降比较明显;而在SBA-15载体上,Pt颗粒本身就容易发生团聚,在反应一开始就表现出非常低的催化性能。此外,载体的孔道结构对反应的选择性、稳定性及结焦也会产生一定的影响[33]。
助剂的添加可以明显改善纯Pt催化剂稳定性差、选择性低等缺陷。Kumar等[12]用湿法浸渍制备了Pt/SBA-15和PtSn/SBA-15催化剂,考察了Sn助剂对丙烷脱氢反应的影响。在Pt催化剂中引入Sn后形成了Pt-Sn合金,提高了Pt颗粒的分散度,从而使PtSn/SBA-15催化丙烷脱氢反应表现出更高的丙烷转化率、丙烯选择性和稳定性。但是,Sn助剂的加入也随之带来一些负面影响,例如催化剂结焦现象加剧,其结焦含量高达Pt/SBA-15催化剂的3倍之多;然而该改性催化剂对积炭具有较强的排出效应,所产生的积炭并没有对活性位产生影响,仍表现出优异的催化性能。助剂Sn对Pt/SAPO-34催化剂结构的影响也非常明显。Nawaz等[13]采用H2-TPR手段对Pt/SAPO-34和PtSn/SAPO-34进行了分析,前者在450~600℃内出现了尖锐的还原峰,而后者在360~540℃内出现了较宽的还原峰,还原温度较前者有明显降低,说明Sn的添加促进了Pt的还原,且Sn和Pt两者间的相互作用有利于脱氢反应的进行。Sn的价态对催化剂的影响也非常大。Sn0与Pt形成Pt-Sn合金,使Pt中毒产生不可逆失活,而Sn2+则提高Pt晶粒分散度,降低氢解活性,对Pt的催化性能具有较大的促进作用。Zhang等[34]进一步研究了Sn含量对PtSn/ZSM-5催化剂活性的影响,发现适量助剂Sn的添加不仅具有“几何效应”,降低了表面Pt粒子的团聚,同时增强了载体与金属之间的相互作用;而且Sn还可以促进积炭由活性位向载体发生转移,提高催化剂的稳定性。但当添加的Sn过量时,会形成更多的Sn0物种,引起催化剂中毒,不利于催化反应的进行。因此在Pt催化剂体系中引入适量的助剂Sn能够有效地提高催化活性和稳定性。
除Sn助剂外,Cu的添加对Pt催化剂的丙烷脱氢催化活性也有明显的促进作用。Pt-Cu/Al2O3催化剂比Pt/Al2O3具有更高的丙烯选择性和更低的失活速率,同时有着更强的抗积炭能力[14]。适量Cu(0.5%,质量分数)的添加可以改变Pt催化剂粒子的形貌和原子的电子环境,Pt-Cu间的相互作用抑制了丙烯的吸附,并提升了C—C键断裂的能垒,从而有效抑制了积炭的产生及裂解反应的发生。适量助剂Cu的引入有效地将积炭由活性位转移到载体上,提高了催化剂的选择性和稳定性。
金属氧化物助剂的添加也在一定程度上可改善Pt系催化剂的活性。Yu等[35]考察了不同金属氧化物(Ce,Sn,V,Fe,Mn,Zr,La,Cr等)助剂对Pt/γ-Al2O3催化丙烷脱氢反应的影响,发现只有SnO2、CeO2和ZrO2这3种助剂可以明显提高催化活性,初始转化率分别提高了8.2%、9.2%和8.1%。
为了进一步提高PtSn负载型催化剂的稳定性和选择性,还可以加入第二种甚至第三种助剂。Yang等[36]在PtSn/γ-Al2O3催化剂中分别添加Mg和K,调变了催化剂的酸碱性,改善了催化剂的丙烷脱氢反应催化性能;将Mg的作用部分归结为稳定了高价锡氧化物,使其不易被还原,当添加过量的Mg时,则促进低价锡氧化物进一步转化为零价锡,从而导致催化剂中毒。Huang等[15]探讨了第二助剂Sr的含量对PtSn/HZSM-5催化剂性能的影响。Sr的加入提高了Pt的分散度,适量的Sr(1.2%,质量分数)可提高催化剂的活性,并有效地减少催化剂的积炭量;再添加第三种助剂Na时,进一步提高了PtSnSr/HZSM-5催化剂的活性,反应5 h后丙烯的选择性大于95%,丙烷转化率为32.2%。引入第三种助剂稀土金属La,中和了Pt催化剂表面的部分弱酸中心和强酸中心,有效地改善了催化剂对产物的选择性和稳定性,同时La的加入可以有效抑制Sn的还原;但过量的La则减少了催化剂表面Pt活性位点数量而导致催化活性下降,积炭量也会相应增加。此外,适量的助剂Ce的加入可以提高Pt的分散度、催化剂的活性和稳定性,且抑制积炭在催化剂表面的聚集[16,37]。由此可见,催化体系中引入适量的助剂能够有效地改善催化剂的催化活性、产物选择性和稳定性。
Pt系催化剂和Cr系催化剂类似,它们都存在稳定性较差的问题。由于Pt和Cr都是金属,含有大量的酸性位,容易导致丙烷的深度脱氢和催化剂表面的结焦而失活。加入碱土金属或富电子的过渡金属,能有效改善催化剂表面的酸性位和电子密度,从而显著提高催化剂的稳定性。但是,Pt的价格昂贵,来源受到限制,难以满足工业生产的要求;另外,催化剂失活仍然是困扰丙烷脱氢行业最重要的问题。
1.3 碳催化剂
近年来,碳材料作为一种新兴的催化剂得到了广泛关注,利用碳材料表面的官能团作为活性位点应用于催化反应的研究也越来越多。其中有序介孔碳材料具有均一的介孔孔径、较高的比表面积和较大的孔容,有可能产生较多的活性中心,并提供分子限域效应,从而有利于多相催化反应中产物分子的选择性生成。在不负载任何金属或者金属氧化物的情况下,纳米和多孔碳材料可以直接作为催化剂应用于碳氢化合物的脱氢反应[38-39]。Liu等[17-18,40]首次将介孔碳材料用于催化丙烷直接脱氢制丙烯的反应,表现出优异的催化活性;反应进行100 h,丙烯的选择性仍可以持续保持在90%左右,丙烯的收率达到38%(见图2),与Pt/Sn-Ce的催化活性相当,且明显高于Pt/Sn基催化剂。XPS O1s能谱的结果表明,介孔碳材料催化剂的丙烷脱氢催化活性与其表面不饱和C=O的含量成正比[17]。反应的机理是,烷烃分子C—H键先在C=O位上进行脱氢反应,同时C=O转变成C—OH。然后,不同反应条件下活性位的再生过程不一样,在氧化脱氢条件下,O2与脱下的H原子反应生成水,C=O活性位得以循环;在直接脱氢反应中,C—OH在高温条件下分解脱去H2,得到再生的C=O活性位。硝酸活化处理后的样品,引入了更多的C=O活性基团,催化活性得到明显提高,但同时也增加了材料表面的酸性基团,引起深度脱氢的发生,导致丙烯的选择性相对较差[18]。此外,介孔碳材料有序的孔道结构可以提高传质作用,同时增强热稳定性,有利于提高催化活性和反应稳定性[41]。
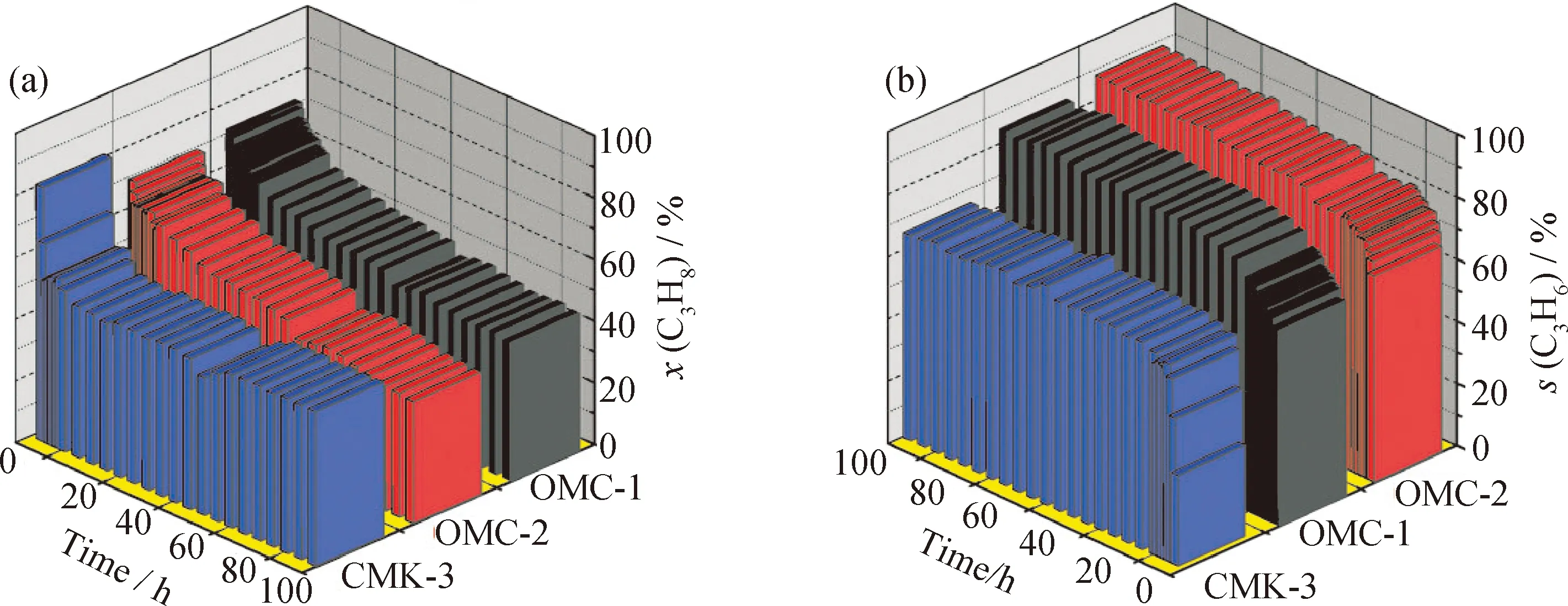
图2 不同碳催化剂催化丙烷脱氢反应的丙烷转化率和丙烯选择性随反应时间的变化
与有序介孔碳材料催化剂相比,活性炭、碳纳米管、石墨等的催化活性和稳定性都很差。活性炭主要是一些微孔结构,不利于反应物与产物的传输;在反应初期,催化活性相对较高,但是随着反应时间的增加,大量的微孔孔道会被积炭堵塞,活性位点也随之减少,因而稳定性较差。石墨碳和碳纳米管则几乎没有活性,因为它们的表面含氧基团量较低,表面活性位点较少,且比表面积很低,活性位点也不容易暴露出来,降低了反应物与活性位点接触机会,导致催化活性很低。
虽然碳材料催化剂和负载型金属催化剂类似,都需要有优良的孔道特性和足够的活性位点,才能使反应物有效地扩散到催化剂内部并吸附到活性位点上,然而有序介孔碳材料具有金属材料不可比拟的性质。一方面,碳材料表面含有大量的C=O基团,这些供电子基团能够有效地与丙烷进行相互作用,从而将丙烷分子的C—H键活化,达到脱除氢的目的;另一方面,由于其优良的孔道特性,反应物和产物分子能够在孔道内部快速传输,实现丙烷的快速活化和丙烯的迅速脱附,从而催化剂稳定性较高不易失活。同时,碳材料具有质量轻、密度小、比表面积大、无污染等特点,在催化方面具有很大的应用前景。尽管有序碳材料尚未商业化,开发方便、快捷的方法合成碳催化剂是面临的一个挑战。
2 丙烷氧气氧化脱氢制丙烯
虽然丙烷直接脱氢已经实现了工业化,但仍然存在不少问题,如反应温度较高,能耗大,催化剂积炭失活快,需要不停地再生处理等。丙烷氧化脱氢是一个放热过程,反应温度相比于直接脱氢明显降低,并且氧气的参与可以减少积炭的产生。因此,丙烷氧气氧化脱氢制丙烯反应已倍受关注。
丙烷在氧气氛中氧化脱氢生成丙烯是一个放热反应,平衡转化率不受热力学限制,一般在400~500℃下进行,而且氧气氛围可以除去催化剂表面积炭,减缓催化剂失活。但是,反应过程中不可避免地会发生丙烷深度氧化。同时,丙烯的C—H键键能小于丙烷的C—H键键能,更容易被深度氧化为碳氧化合物。在许多催化剂体系中,烷烃中第1个H的消除是速控步骤。由于这一步反应的能垒较高,需要催化剂具有一定的氧化能力,这同时会引起丙烷和丙烯的深度氧化,因而烷烃的活化条件相当苛刻。
关于丙烷氧气气氛下氧化脱氢的机理主要有2个观点。一种是自由基反应机理,另一种是Mars-Van Kervelen机理,即氧化还原机理。2种反应机理存在的条件不同,自由基反应主要存在于气相中,而氧化还原主要存在于催化剂的表面。
自由基反应机理如式(5)~式(9)所示。
C3H8+O2→C3H7·+HO2·
(5)
C3H7·+O2→C3H6+HO2·
(6)
C3H8+HO2·→C3H7·+H2O2
(7)
H2O2→2OH·
(8)
C3H8+OH·→C3H7·+H2O
(9)
Michalakos等[42]认为,在自由基反应机理中,丙烷亚甲基上的C—H键断裂形成吸附态的丙基自由基,接着终端C—H键断裂形成丙烯,这其中形成丙基自由基的步骤是速控步。在该反应过程中,还存在其它的链增长和链终止步骤,生成H2、CH4、CO、CO2和H2O等。丙烷分子中C—H键断裂分为均裂和异裂,即均裂生成丙基自由基(CH3CH·CH3),异裂生成碳正离子(CH3C+CH3)或碳负离子(CH3C-HCH3)[43]。反应过程中碳正离子在酸性催化剂上出现,而碳负离子在碱性较强的催化剂上生成。
氧化还原(Mars-Van Krevelen)机理如图3所示。烷烃分子首先被催化剂表面的氧化活性位氧化,得到脱氢产物的中间体,然后进一步转化为产物,而催化剂则被还原;被还原的催化剂由氧气或吸附态氧迅速补充失去的晶格氧而得到再生[44-45]。氧化还原机理特别适合用于过渡金属氧化物催化剂上的烷烃氧化反应。
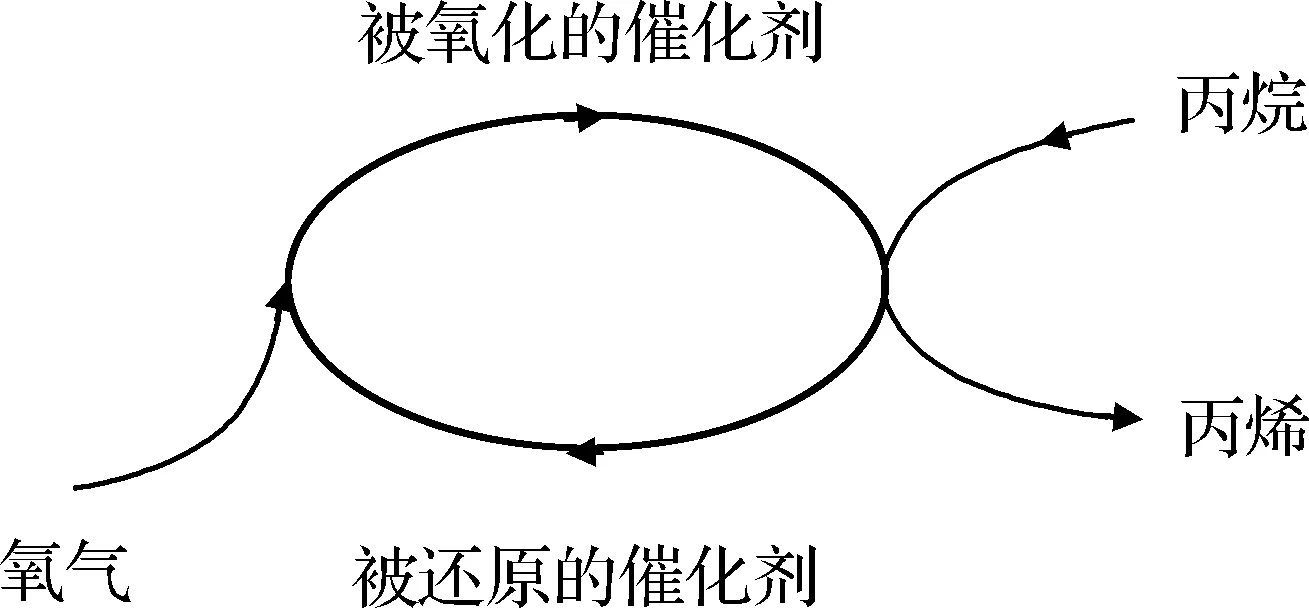
图3 Mars-Van Krevelen过程示意图
丙烷氧气氧化脱氢制丙烯反应主要的催化剂有钒基催化剂、钼基催化剂、稀土基催化剂以及碳催化剂。表3列出了一些常见的丙烷氧气氧化脱氢制丙烯催化剂的制备方法及催化性能。
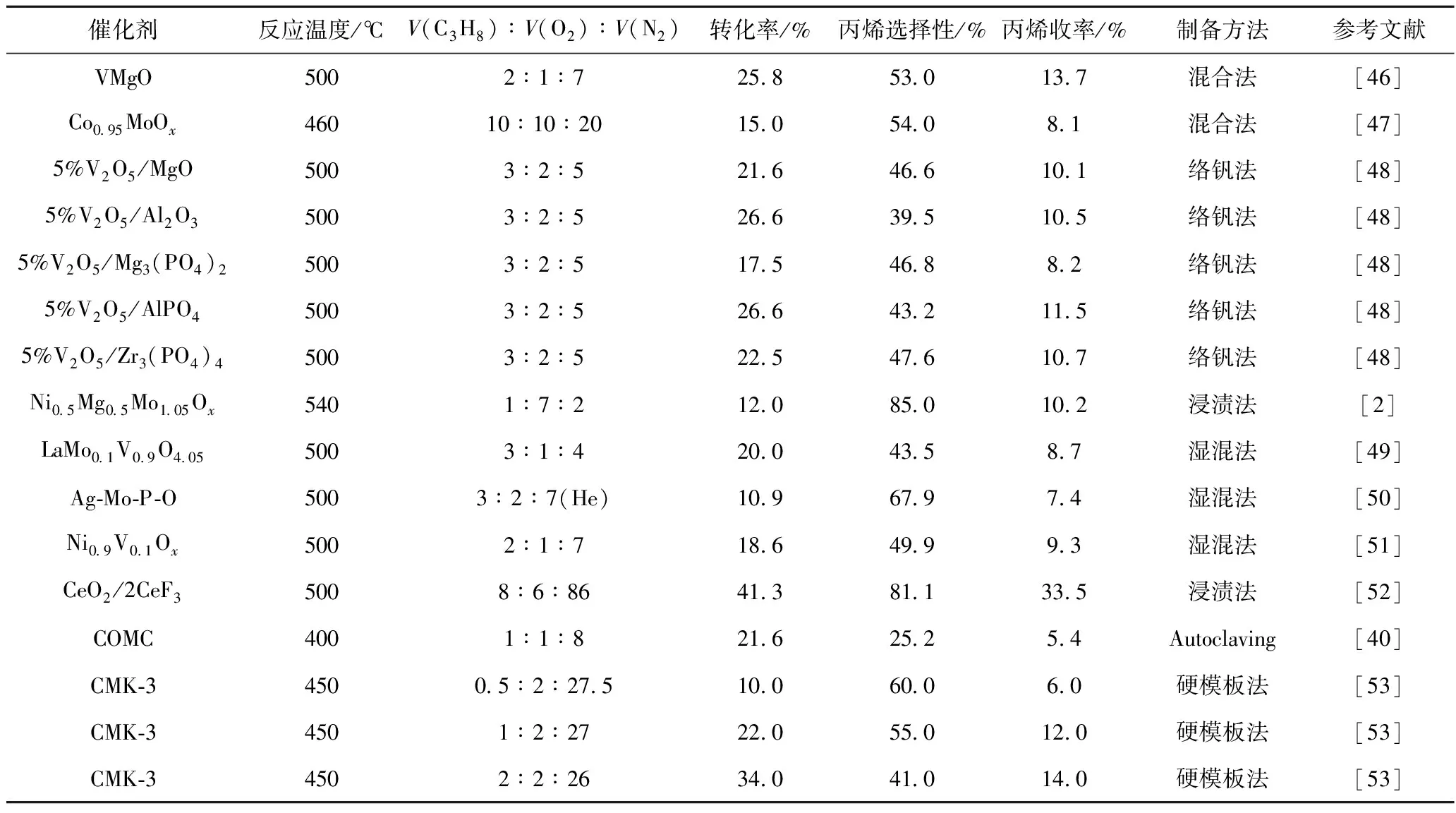
表3 常见丙烷氧气氧化脱氢制丙烯催化剂的催化性能
2.1 钒系催化剂
钒系催化剂由于其独特的电子结构,在丙烷氧化脱氢反应中表现出优异的催化性能。常见的钒系催化剂可以分为VMgO催化剂和负载钒基催化剂。
VMgO催化剂在丙烷氧化脱氢反应中具有较高的催化活性和丙烯选择性。高温焙烧不同化学计量比的V2O5和MgO的混合物,可以得到不同相的VMgO催化剂。VMgO催化剂以正钒酸镁Mg3V2O8、焦钒酸镁α-Mg2V2O7和偏钒酸镁MgV2O63种稳定相化合物形式存在。以这3种纯相作为丙烷氧化脱氢反应催化剂时,Mg3V2O8得到的丙烯选择性最高,α-Mg2V2O7次之,MgV2O6的丙烯选择性和活性都比较差[54]。由于V物种的聚积状态不同,催化活性也有差异,对该催化剂活性相和作用机理也存在着争议。Kung等[55]认为,Mg3V2O8是活性相,丙烷先后断裂亚甲基和甲基上的C—H键生成丙烯,催化剂的碱性有利于丙烯分子的脱附,且Mg3V2O8表面不存在V=O键从而使产物中无含氧有机物生成。Volta等[56]认为活性相是α-Mg2V2O7,该相中稳定存在着与氧缺位形成相关的V4+,表面存在能够脱氢的V=O键和同时生成水的共角VO4四面体,反应生成少量的含氧有机物。他们认为该体系反应属于Mars-Van-Krevelen类型的反应机理,但无法确定非分子氧的来源是体相氧还是化学吸附氧。Fang等[57]则认为含适量V4+的钒酸镁才可能是活性相。这主要是由于不含V4+的钒酸镁具有很规整的晶体结构和较差的氧化还原性,反应过程中V5+不能被还原为V4+,不能有效地形成V4+/V5+氧化还原偶,而含有V4+的钒酸镁具有更好的氧化还原性,V4+不仅能与邻近的V5+构成氧化还原偶,而且还可能与更低价的V3+或V2+构成氧化还原偶,有效促进反应的进行。他们进一步证明了VMgO催化剂催化丙烷氧化脱氢反应按Mars-Van-Krevelen晶格氧反应机理进行[46],认为丙烷的活化需要表面晶格氧物种;这些氧物种一旦生成就能很快与丙烷作用,且氧的吸附活化并转化为晶格氧是该反应过程的重要步骤。
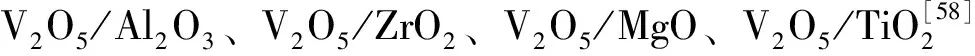
钒负载量对负载型钒催化剂的催化丙烷氧化脱氢活性影响非常大,单层负载量的催化剂显示出了很高的烯烃选择性。因为负载量不同,在催化剂表面形成的活性物质种类也不一样,表现出不同的催化活性。在低钒负载量时,催化剂表面主要是孤立的单分散氧化钒物种,随着钒负载量的增加,逐渐由聚合态的氧化钒变成块状的V2O5。V—O—V和V=O被认为是选择氧化的活性物种,同时它们也会对丙烯深度氧化。Chen等[48,67]认为,在钒基催化剂中,载体的金属离子与钒氧化物形成了V—O—M桥键,该桥键氧较易移动,可能是丙烷氧化脱氢反应的活性氧物种。聚合态的氧化钒相比于孤立单分散的氧化钒具有更好的催化活性,表面存在的V4+/V5+为活性中心,保持催化剂上氧化还原反应的循环。所以,要得到较高催化活性的钒基催化剂,应该合理控制其钒负载量。
无论是负载型还是非负载型的钒基催化剂,钒的存在状态和V=O键对丙烷氧化脱氢过程影响很大。但是对于钒基催化剂的活性位点还存有很大争议,仍有大量的问题值得深入探讨。
2.2 钼系催化剂
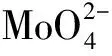
多组分钼基催化剂较纯相氧化物显示出了更高的丙烷氧化脱氢反应催化活性,如Ni-Mo-O、Mg-Mo-O、Co-Mo-O等以及负载在Al2O3、ZrO2、SiO2、Nb2O5、TiO2、MgO上的钼基催化剂。Ni-Mo-O催化剂中,表面的O2-被认为是反应的活性物种[71]。一般在钼基催化剂中含有MoOx和其它的混合相,MoOx团簇被认为是丙烷氧化脱氢反应的活性位和选择相[70,72]。在Co-Mo-O体系中,当Mo稍过量时,催化剂的活性和丙烯选择性很高,归因于少量的MoO3覆盖了表面相对较强的酸性位,提供了弱酸酸位[47],而较弱的酸性位可能更有利于提高催化剂对该反应的催化活性和选择性。
钼基催化剂中,添加一定组分的助剂也可以改善其催化活性。在Mo—V—O中,Co的添加提升了催化剂的丙烷氧化脱氢性能[73]。XRD表征表明,催化剂由原先的Mo6V9O40和MoO3两相转变成Mo4V6O5和CoMoO4两相,这可能是催化活性提高的一个主要原因。TPR分析证明,Co的添加提高了钼基催化剂的还原性,有利于晶格氧的迁移和插入,促进了丙烷的活化,进而提高了丙烷的转化率。NH3-TPD进一步证明了Co添加后的钼基催化剂的酸性降低,解释了丙烯选择性提高的原因。
2.3 稀土催化剂
稀土催化剂也被广泛用于丙烷氧化脱氢制丙烯反应。LuVO4对该反应具有良好的催化效果,其催化性能也因制备的方法不同而变化[74-75],过渡金属的添加也可以提高丙烯的选择性。助剂的含量、前驱物以及制备方法等对LuVO4催化丙烷氧化脱氢的活性和选择性都会产生一定的影响,因为助剂与LuVO4的相互作用抑制了氧化能力强的活性位,减缓了丙烯的深度氧化。该体系中,与氧缺位形成有关的V4+是活化氧分子的活性中心[76],与V—Mg—O复合氧化物催化剂的活性相一致[46]。
稀土基氟化物对丙烷氧化脱氢制丙烯反应也具有很有效的催化性能[52,77]。3%Cs2O/2CeO2/CeF3催化丙烷氧化脱氢反应的丙烷转化率和丙烯选择性分别高达53.4%和67.5%。由于CeF3的加入使得稀土氧化物中的晶格O2-被F-交换,形成了氧缺位;同时,表面的F-可以分隔催化剂表面的活性氧物种,避免了丙烷和丙烯的深度氧化,提高了丙烯选择性。
稀土金属氧化物催化剂在催化丙烷氧化脱氢反应中具有很多其他金属不可比拟的优势,但由于其价格昂贵,很难实现工业化。而且,稀土作为一种重金属,使用过程中不可避免地会对环境产生一定的污染,因此有待进一步开发更为环保、高效的催化剂。
2.4 碳基催化剂
近年来,纳米碳材料由于其独特的电子特性和丰富的表面官能团越来越引起广泛关注。Su等[38,78-79]对于纳米碳材料在催化脱氢方面的性能进行了大量的研究工作。碳材料表面含有丰富的含氧官能团,包括酸性官能团(—COOH,—C—OH)和碱性官能团(C=O),在脱氢反应中表现出很大优势。Qi等[80]首次采用化学滴定的方法对碳材料表面不同的含氧官能团进行选择性失活,并对其定性和定量分析。经过苯肼溶液处理的碳纳米管表面的C=O基团消失,在催化乙苯氧化脱氢的实验中催化活性明显下降,有力地证明了碳材料表面的C=O基团为脱氢反应的活性位点。
有序介孔碳由于具有均一的介孔孔道结构和表面丰富的C=O官能团,在催化丙烷直接脱氢时显示出优越的催化活性和稳定性。同样的催化剂在氧气气氛下催化丙烷脱氢时,其丙烯选择性则非常差,只有25.2%,导致最终的丙烯产率仅为5%左右[40]。这是由于高比表面积的介孔碳在催化丙烷脱氢过程中会将目标产物丙烯再次吸附在其表面,导致丙烯过度脱氢和深度氧化,使丙烯选择性降低。Piotr等以SBA-15为硬模板、蔗糖为碳源,合成了CMK-3介孔碳,进一步讨论了反应物中氧气的含量对催化丙烷氧化脱氢活性的影响[53]。随着氧气分压的增加,丙烷的转化率随之升高,但丙烯选择性则逐渐降低。在稳定性试验中,随着反应时间的延长,丙烯收率呈下降趋势,且氧气含量越高,下降趋势越明显。在该反应过程中,部分碳基催化剂本身与氧气发生了反应,因此导致催化活性的降低。
除了介孔碳以外,杂原子掺杂的碳纳米管也可用于催化丙烷氧化脱氢反应。Chen等[81]合成了氮掺杂的碳纳米管,在催化丙烷氧化脱氢反应中显示出比未掺杂的碳管更高的催化活性和稳定性,且丙烷转化率和丙烯选择性随氮含量的增加而增加。掺氮后碳管有利于电子传递,对氧分子的吸附量更大,解离吸附更容易,有利于表面的—OH物种氧化再生为C=O活性中心,从而促进了丙烷氧化脱氢反应的进行。另一方面,氮对富电子烯烃有排斥作用,使其更容易从表面脱附,减少被深度氧化的几率,提高丙烯选择性。因此,对于杂原子改性的碳材料可能是该反应体系未来的一个发展方向。
目前,碳材料用于丙烷氧化脱氢反应的研究并未成熟,对于其机理性探究还有待进一步深入。同时,碳基催化剂也存在着积炭堵塞催化剂孔道的问题,如何对积炭进行消除,目前还未见报道。此外,人工合成的碳基催化剂价格昂贵,很难大规模生产,距离工业化还比较遥远。
3 丙烷二氧化碳气氛氧化脱氢
丙烷氧气氧化脱氢已经克服了直接脱氢反应的缺点,在较低温度下就可以获得较高的丙烷转化率。但是,氧气是一种强氧化剂,对产物发生深度氧化,导致丙烯选择性降低。二氧化碳可作为一种弱的氧化剂用于丙烷脱氢反应,一方面缓解了丙烷深度氧化的问题,另一方面还能减少温室气体的排放,对于环境保护具有非常重要的意义。
丙烷在二氧化碳气氛中的脱氢反应存在“一步法”和“两步法”两个可能的脱氢过程。
“一步法”脱氢过程遵循Mars-Van-Krevelen机理。催化剂表面活性物种的晶格氧用于转化烃类物种,生成目标产物烯烃和水,同时二氧化碳再为催化剂表面补充晶格氧,并释放一氧化碳(C3H8+CO2→C3H6+H2O+CO)。以Cr基催化剂为例的反应机理如图4所示。该反应过程中,晶格氧所发挥的作用与碳基催化剂临氧脱氢中的分子氧类似,与脱附的H结合生成产物H2O;CO2则起到弱氧化剂的作用,将还原态的Cr(Ⅲ)进一步氧化为Cr(Ⅵ),实现了催化剂循环再生的过程。
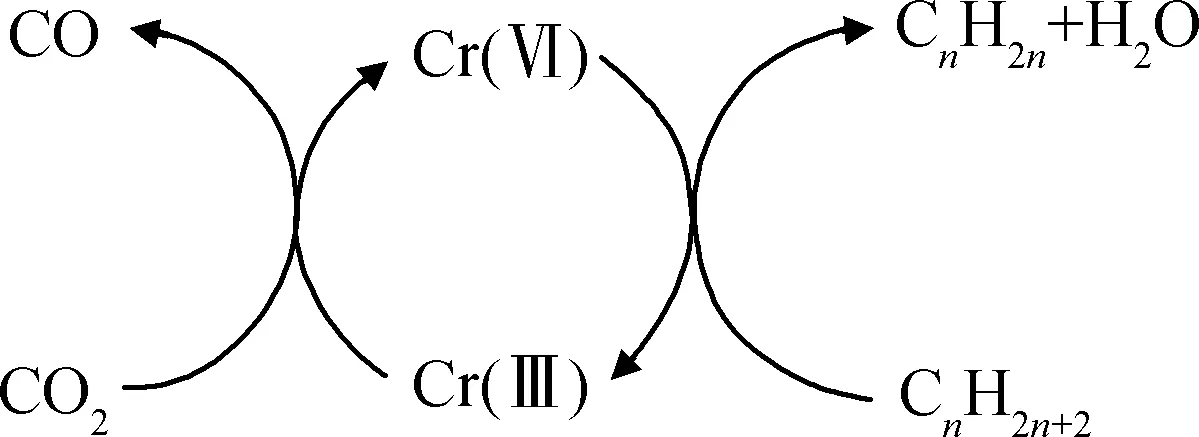
图4 临CO2条件下Cr基催化剂催化丙烷脱氢反应机理
对于“两步法”脱氢过程,首先丙烷分子在催化剂表面发生脱氢反应,生成H2,然后H2和CO2再通过逆水煤气变换反应(RWGS)生成H2O和CO,如式(10)、式(11)所示。
C3H8→C3H6+H2
(10)
H2+CO2→H2O+CO
(11)
“两步法”机理中,第一步反应类似于直接脱氢过程,第二步则利用CO2通过RWGS反应将产生的H2迅速消除,有利于平衡向正方向进行。另外,还可以通过CO2+C→CO反应除去产生的积炭,提高该反应体系的稳定性。
丙烷二氧化碳气氛氧化脱氢制丙烯反应主要的催化剂有铬基催化剂和镓基催化剂,常用的催化剂的制备方法及催化性能列于表4。
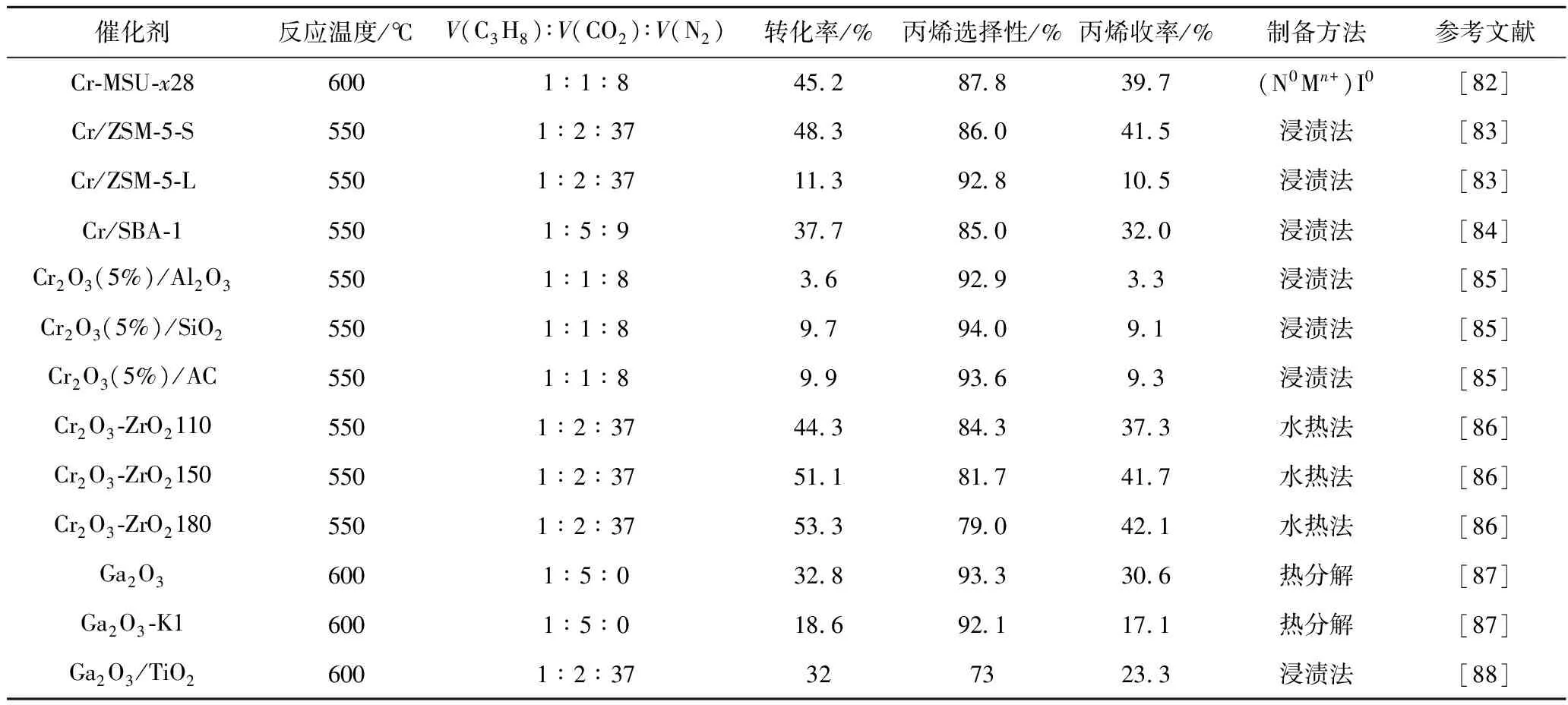
表4 丙烷二氧化碳气氛氧化脱氢制丙烯催化剂的催化性能
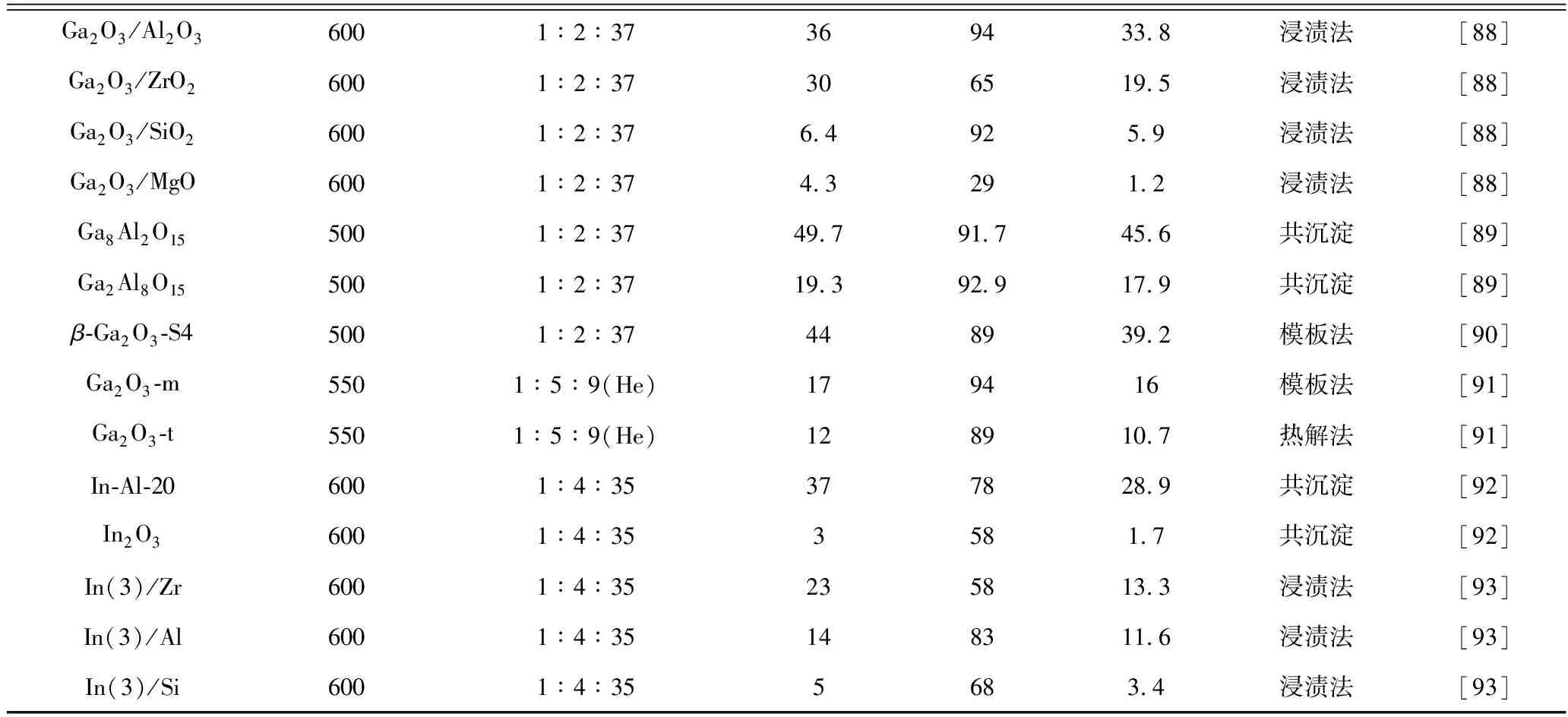
续表4
3.1 铬基催化剂
Cr基催化剂在丙烷直接脱氢反应中表现出非常高的催化活性,但是失活非常严重,一般反应2 h活性会降低到原来的50%,因而需要反复再生;如果能在弱氧化剂的条件下进行脱氢反应,就可以有效抑制积炭的发生,提高催化剂的使用寿命。Takahara等[85]将Cr2O3负载在Al2O3、SiO2和活性碳等不同载体上,比较了这些催化剂在CO2气氛和直接催化丙烷脱氢的活性。结果表明,Cr2O3/Al2O3催化剂对丙烷CO2气氛脱氢的催化活性比直接脱氢要低;Cr2O3/活性炭的两类脱氢催化活性相当;而对于Cr2O3/SiO2催化剂,丙烷转化率由直接脱氢时的6.5%上升到CO2气氛中的9.1%,且催化剂的稳定性明显提高。Cr2O3/SiO2作为催化剂时,CO2可以氧化部分被还原的Cr物种,进而抑制了催化剂的失活,提高了催化剂稳定性;而氧化铝载体的酸碱性随CO2的引入而发生改变,对催化活性极为不利。这也表明,在CO2气氛下载体对催化活性的影响非常大,硅基材料成为该催化剂体系最常用的载体。
除了催化剂载体以外,Cr基催化剂体系中活性位的探讨依然是热点[82-84,94-95]。Takehira等[94]采用UV-vis、UV-Raman、XANES和EXAFS方法表征Cr-MCM-41催化剂,证明了孤立态四面体配位的Cr(Ⅵ)为主要的活性中心,反应过程中被还原为低活性的聚合态八面体配位的Cr(Ⅲ)。还原态的八面体Cr(Ⅲ)O6可以通过O2或CO2再次氧化为四面体Cr(Ⅵ)O4。Cr(Ⅲ)O6和Cr(VI)O4间的氧化还原循环在催化丙烷脱氢反应中起到重要作用。
Cr的负载量对催化剂中Cr的存在形式的影响也很大,不同含量Cr负载到Al2O3时Cr的价态分布列于表6[95]。同样,在硅基载体上Cr的存在形式也会随负载量变化而改变。Michorczyk等[84]制备了一系列Crx/SBA-1催化剂,并研究了Cr的存在状态。结果表明,Cr的存在状态主要包括Cr6+、分散的Cr5+和Cr2O3晶体。Cr6+物种主要以单体或二聚的形式存在,与Cr的含量无关;Cr5+主要在Cr质量分数低于7%时出现;而α-Cr2O3晶体则存在于Cr质量分数大于5%的催化剂中。当Cr质量分数为7%时,Cr催化剂的催化活性最高;进一步增加Cr的负载量,催化活性并没有提高,因为大量的Cr主要以不活泼的Cr2O3晶体形式存在。Operando UV-vis DRS测试结果表明,在反应初始阶段,Cr6+迅速还原为Cr2+/Cr3+,Cr2+和Cr3+与Cr6+间的氧化还原循环成为主要的反应活性中心。此外,Cr(Ⅵ)的形态与催化活性也有关系。Jayeon等[82]将孤立的和聚合的Cr6+分别定义为硬Cr6+和软Cr6+,且两者具有不同的还原性能,软Cr6+比硬Cr6+更容易还原。由软Cr6+还原成的配位不饱和Cr3+的催化活性比硬Cr6+还原得到的Cr3+要高,因为软Cr6+还原得到的Cr3+物种促进了逆水煤气转换反应。另外,催化剂的合成方法对催化活性的影响也很大,Wu等[86]比较了水热法和传统的沉淀法制备的Cr2O3-ZrO2催化剂用于临CO2丙烷脱氢的催化效果,180℃水热处理后的样品的催化活性是沉淀法的1.6倍,主要归因于水热法可以产生更高浓度的Cr6+物种。直接脱氢和临CO2脱氢反应的主要的活性位存在着明显的区别,Cr3+被多数研究者认为是直接脱氢中的主要活性位,虽然在初始反应阶段由于Cr6+的存在表现出高的催化活性,但是Cr6+在高温下很容易被反应气体还原为Cr3+,且没有氧化剂进一步将其氧化为Cr6+,导致活性降低。然而在CO2气氛下催化丙烷脱氢的活性位则是Cr6+物种,在反应过程中Cr3+可以被CO2再次氧化为Cr6+,另一方面CO2的存在抑制了积炭的产生,所以Cr催化剂在CO2气氛下催化丙烷脱氢展示出更优的催化性能。但是,CO2是一种温和的弱氧化剂,只能将少部分易氧化的Cr3+氧化为Cr6+,因此对催化活性提高有限,如何进一步提高对Cr3+的氧化也是值得探讨的问题。
3.2 镓基催化剂
1998年,日本科学家Nakagawa首次报道了镓基催化剂在脱氢反应的应用[96],此后Ga2O3催化剂引起了研究者们的广泛兴趣。Michorczyk等[97]比较了873 K、CO2气氛下不同金属氧化物催化剂催化丙烷脱氢反应,Ga2O3表现出最高的初始活性,反应0.17 h时丙烷转化率约为30%,而Fe2O3和Cr2O3的活性都不高,转化率不到10%。当这些金属氧化物负载到载体上后,催化活性则发生了改变。反应0.75 h时,Cr2O3/Al2O3催化丙烷脱氢反应的转化率为12.7%,而Ga2O3/Al2O3催化丙烷脱氢反应的转化率仅5.4%,这可能是由于活性组分与载体间相互作用不同的缘故。反应温度也是影响催化活性的一个重要因素。Michorczyk等[87]考察了不同反应温度对CO2气氛下Ga2O3催化丙烷脱氢活性的影响,测试温度从673逐步提高到873 K;当温度超过873 K时,丙烯的选择性下降和积炭量增加的现象都迅速加剧,所以适合Ga2O3脱氢反应的温度在773~823 K之间。此外,Ga2O3的晶相对催化活性的影响也很明显。Zheng等[98]考察了α,β,γ和δ4种晶型的Ga2O3在CO2气氛下催化丙烷脱氢反应的活性,其中γ-Ga2O3催化剂的表观活性最好;但由于γ相的比表面积大于β相,根据比表面归一化后的Ga2O3本征活性却是β相的最高,从而认为高的活性与β相表面活性Ga物种的密度最高有关。载体也是影响催化剂活性的关键因素。Xu等[88]继而研究了β-Ga2O3负载于不同氧化物载体上的催化剂用于丙烷的临CO2脱氢反应。通过对TiO2、ZrO2、Al2O3、SiO2和MgO 5种载体的比较发现,载体的酸性对Ga2O3脱氢活性影响显著,其中酸性较强的TiO2、ZrO2、Al2O3负载的Ga2O3催化剂活性明显优于SiO2和MgO负载的Ga2O3催化剂,而且采用TiO2为载体的Ga2O3催化剂时,CO2通过逆水煤气转化反应以提高丙烷转化的效果最明显,ZrO2和Al2O3为载体时的Ga2O3催化剂在有CO2气氛下的初始活性反而不如在无CO2时的初始活性高。
除了负载型的Ga2O3催化剂,Ga基固溶体也是一类重要的CO2气氛下丙烷脱氢催化剂[89,99]。Chen等[89]通过乙醇-氨水共沉淀法制备出了一系列不同Ga/Al摩尔比的GaxAl10-xO15固溶体催化剂,催化丙烷脱氢的结果表明,Ga2O3-Al2O3固溶体稳定性明显高于Ga2O3。Al2O3的加入使得到的固溶体中四配位的Ga(Ⅵ)物种质量分数上升,增加了弱Lewis酸位,而这种弱酸位对催化活性的提高有很重要的作用。另外,丙烯在Ga2O3-Al2O3固溶体表面比Ga2O3表面更容易脱附,使得催化剂积炭更少,稳定性更高。
Ga2O3催化剂的孔结构也是影响其催化活性的一个重要方面。以SBA-15为硬模板合成的有序介孔结构Ga2O3-m催化剂比传统热分解法制得的Ga2O3-t催化剂具有更高的结构稳定性和抗失活性能[91]。蔗糖作为一种环境友好型的模板剂,也可以用来合成介孔β-Ga2O3催化剂,且催化活性比Ga2O3-m更高。主要是该催化剂具有有利的结构特征和高的比表面积使得其含有大量配位不饱和的表面Ga活性位,从而使催化性能得到提高[90]。在Ga基催化剂体系,固溶体及介孔结构催化剂具有更优越的催化活性,如果将两者相结合,合成具有介孔结构的镓-铝固溶体催化剂,将有可能进一步提高其催化性能。
3.3 铟基催化剂
与Ga同一主族的In基催化剂也可被用于在CO2气氛中催化丙烷脱氢反应。Chen等[92]通过共沉淀法制备了In2O3-Al2O3催化剂,比较了In/Al摩尔比、反应温度及CO2分压对催化活性的影响。复合氧化物In2O3-Al2O3的脱氢活性远高于单组分氧化物,在873 K和10 kPa CO2条件下,丙烷的转化率可以达到35.7%,丙烯选择性为76.5%。原位生成的In0是脱氢活性中心,而体相的In2O3则是逆水煤气转换的活性中心,两者相互作用共同促进丙烷脱氢催化反应的进行。另外,载体也是影响In基催化剂活性的一个重要方面。Chen[93]等将In2O3负载在SiO2、Al2O3和ZrO23种载体上,并对所得催化剂的结构、氧化还原性质和酸碱性进行了表征。结果表明,In2O3在Al2O3和ZrO2上的分散性相对较好,在SiO2上的分散性相对较差,且各催化剂的酸碱性差别也很大。在CO2气氛下丙烷脱氢的活性从大到小的In基催化剂的顺序为In/Zr、In/Al、In/Si。载体对In2O3分散能力越好的催化剂越有利于产生更多的活性In0物种,且碱密度越高越有利于逆水煤气转换发生,进而促进脱氢反应的进行。目前,对于In基催化剂用于催化丙烷脱氢的报道还不是很多,同时有些问题尚待解决。比如,如何在对该催化剂进行再生去除积炭时保持In0不被氧化,如何在制备高负载量催化剂时使活性物种具有更高的分散性等。
4 总结与展望
丙烷脱氢制丙烯技术是解决丙烯持续增长需要的一条重要途径,具有应用前景。丙烷直接脱氢生产丙烯已经实现了工业化,但丙烷转化率受热力学平衡限制而难以提高,而且催化剂失活很快,再生频繁,能量消耗大;丙烷氧气氧化脱氢克服了直接脱氢的热力学平衡的限制,且临氧条件下催化剂表面的积炭容易烧除,抑制了催化剂结焦失活问题,但丙烯会发生深度氧化,降低了丙烯选择性;在丙烷二氧化碳气氛氧化脱氢中,CO2作为一种温和氧化剂,将丙烷脱氢与逆水煤气变换耦合,通过CO2除去H2提高丙烷脱氢反应活性,副产物比氧气脱氢明显减少,但丙烷转化率普遍较低。不同条件下的丙烷脱氢制丙烯反应存在着各自的优、缺点,同时在整个丙烷脱氢反应及催化剂体系仍存在着许多理论问题亟待解决,如反应机理、反应动力学、如何获得合适的载体、如何克服积炭的困扰提高催化剂稳定性等。这些问题的深入研究将对改进催化剂的性能及优化操作条件提供重要指导。
[1] 吴锁林. 丙烷脱氢制丙烯的技术进展[J]. 江苏化工, 1998, 26(2): 33-35. (WU Suolin. Technologic advances preparing propylene by propane dehydrogenation[J]. Jiangsu Chemical Industry, 1998, 26(2): 33-35)
[2] 张伟德, 李基涛, 付锦坤, 等. 丙烷在Ni-Mg-Mo-O催化剂上的氧化脱氢[J]. 天然气化工, 2000, 25(4): 1-4. (ZHANG Weide, LI Jitao, FU Jinkun, et al. Oxidative dehydrogenation of propane over Ni-Mg-Mo-O catalysts[J]. Natural Gas Chemical Industry, 2000, 25(4): 1-4)
[3] 陈建九, 史海英, 汪泳. 丙烷脱氢制丙烯工艺技术[J]. 精细石油化工进展, 2000, 1(12):23-28. (CHEN Jianjiu, SHI Haiying, WANG Yong. Process of producing propylene by propane dehydrogenation[J]. Advances in Fine Petrochemicals, 2000, 1(12): 23-28)
[4] CHEN K, IGLESIA E, BELL A T. Kinetic isotopic effects in oxidative dehydrogenation of propane on vanadium oxide catalysts[J]. Journal of Catalysis, 2000, 192(1): 197-203.
[5] 董文生, 王心葵, 彭少逸. 丙烷脱氢制丙烯研究进展[J]. 合成化学, 1997, 5(3):246-250. (DONG Wensheng, WANG Xinkui, PENG Shaoyi. New progress in propane dehydrogenation to propene[J]. Chinese Journal of Synthetic Chemistry, 1997, 5(3): 246-250)
[6] 余长林. 丙烷脱氢铂催化剂与反应性能的研究[D]. 大连:大连化学物理研究所, 2007: 16-17.
[7] 陈光文, 阳永荣, 戎顺熙. 在Pt-Sn/Al2O3催化剂上丙烷脱氢反应动力学[J]. 化学反应工程与工艺, 1998, 14(2): 130-137. (CHEN Guangwen, YANG Yongrong, RONG Shunxi. Study on the intrinsic kinetics of propane dehydrogenation over Pt-Sn/Al2O3catalyst[J]. Chemical Reaction Engineering and Technology, 1998, 14(2): 130-137.)
[9] 韩玉庭. 丙烷脱氢制丙烯催化剂的研究[D]. 天津: 天津大学, 1999.
[10] SANTHOSH KUMAR M, HAMMER N, RΦNNING M, et al. The nature of active chromium species in Cr-catalysts for dehydrogenation of propane: New insights by a comprehensive spectroscopic study[J]. Journal of Catalysis, 2009, 261(1): 116-128.
[11] 张一卫, 周钰明, 邱安定, 等. Na对PtSn/ZSM5催化剂丙烷脱氢反应性能的影响[J]. 物理化学学报, 2006, 22(6): 672--678. (ZHANG Yiwei, ZHOU Yuming, QIU Anding, et al. Effect of Na addition on catalytic performance of PtSn/ZSM-5 catalyst for propane dehydrogenation[J]. Acta Phys-Chim Sin, 2006, 22(6): 672-678.)
[12] SANTHOSH KUMAR M, CHEN D, HOLMEN A, et al. Dehydrogenation of propane over Pt-SBA-15 and Pt-Sn-SBA-15: Effect of Sn on the dispersion of Pt and catalytic behavior[J]. Catalysis Today, 2009, 142(1): 17-23.
[13] NAWAZ Z, TANG X, ZHANG Q, et al. SAPO-34 supported Pt-Sn-based novel catalyst for propane dehydrogenation to propylene[J]. Catalysis Communications, 2009, 10(14): 1925-1930.
[14] HAN Zhiping, LI Shuirong, JIANG Feng, et al. Propane dehydrogenation over Pt-Cu bimetallic catalysts: The nature of coke deposition and role of copper[J]. Nanoscale, 2014, 6(17): 10000-10008.
[15] LI H, SHIJIAN Z, YUMING Z, et al. Effect of strontium addition to platinum catalyst for propane dehydrogenation[J]. China Petroleum Processing and Petrochemical Technology, 2012, 14(3): 75-82.
[16] YU C, GE Q, XU H, et al. Effects of Ce addition on the Pt-Sn/γ-Al2O3catalyst for propane dehydrogenation to propylene[J]. Applied Catalysis A: General, 2006, 315(1): 58-67.
[17] LIU L, DENG Q F, AGULA B, et al. Ordered mesoporous carbon catalyst for dehydrogenation of propane to propylene[J]. Chemical Communications, 2011, 47(29): 8334-8336.
[18] LIU L, DENG Q F, LIU Y P, et al. HNO3-activated mesoporous carbon catalyst for direct dehydrogenation of propane to propylene[J]. Catalysis Communications, 2011, 16(1): 81-85.
[19] GRÜNERT W, SAFFERT W, FELDHAUS R, et al. Reduction and aromatization activity of chromia-alumina catalysts: I Reduction and break-in behavior of a potassium-promoted chromia-alumina catalyst[J]. Journal of Catalysis, 1986, 99(1): 149-158.
[20] HARDCASTLE F D, WACHS I E. Raman spectroscopy of chromium oxide supported on Al2O3, TiO2and SiO2: A comparative study[J]. Journal of Molecular Catalysis, 1988, 46(1): 173-186.
[21] SOKOLOV S, STOYANOVA M, RODEMERCK U, et al. Comparative study of propane dehydrogenation over V-, Cr-, and Pt-based catalysts: Time on-stream behavior and origins of deactivation[J]. Journal of Catalysis, 2012, 293: 67-75.
[22] PUNRUNEN R L, WECKHUYSEN B M. Spectroscopic study on the irreversible deactivation of Chromia/alumina dehydrogenation catalysts[J]. Journal of Catalysis, 2002, 210(2): 418-430.
[23] CAVANI F, KOUTYREV M, TRIFIRO F, et al. Chemical and physical characterization of alumina-supported chromia-based catalysts and their activity in dehydrogenation of isobutene[J]. Journal of Catalysis, 1996, 158(1): 236-250.
[24] DE ROSSI S, PIA CASALETTO M, FERRARIS G, et al. Chromia/zirconia catalysts with Cr content exceeding the monolayer: A comparison with chromia/alumina and chromia/silica for isobutane dehydrogenation[J]. Applied Catalysis A: General, 1998, 167(2): 257-270.
[25] HAKULI A, KYTÖKIVI A, KRAUSE A O I. Dehydrogenation ofi-butane on CrOx/Al2O3catalysts prepared by ALE and impregnation techniques[J]. Applied Catalysis A: General, 2000, 190(1): 219-232.
[26] HAKULI A, KYTÖKIVI A, KRAUSE A O I, et al. Initial activity of reduced chromia/alumina catalyst inn-butane dehydrogenation monitored by on-line FT-IR gas analysis[J]. Journal of Catalysis, 1996, 161(1): 393-400.
[27] DEROSSI S, FERRARIS G, FREMIOTTI S, et al. Propane dehydrogenation on chromia/silica and chromia/alumina catalysts[J]. Journal of Catalysis, 1994, 148(1): 36-46.
[28] AIRAKSINEN S M K, KANERVO J M, KRAUSE A O I. Deactivation of CrOx/Al2O3catalysts in the dehydrogenation ofi-butane[J]. Studies in Surface Science and Catalysis, 2001, 136: 153-158.
[29] GORRIZ O F, CADUS L E. Supported chromium oxide catalysts using metal carboxylate complexes: Dehydrogenation of propane[J]. Applied Catalysis A: General, 1999, 180(1): 247-260.
[30] CIMINO A, CORDISCHI D, DE ROSSI S, et al. Studies on chromia zirconia catalysts III Propene hydrogenation[J]. Journal of Catalysis, 1991, 127(2): 777-787.
[31] SANTHOSH KUMAR M, CHEN D, WALMSLEY J C, et al. Dehydrogenation of propane over Pt-SBA-15: Effect of Pt particle size[J]. Catalysis Communications, 2008, 9(5): 747-750.
[32] ZHANG Y, ZHOU Y, SHI J, et al. Comparative study of bimetallic Pt-Sn catalysts supported on different supports for propane dehydrogenation[J]. Journal of Molecular Catalysis A: Chemical, 2014, 381: 138-147.
[33] SANTHOSH KUMAR M, HOLMEN A, CHEN D. The influence of pore geometry of Pt containing ZSM-5, beta and SBA-15 catalysts on dehydrogenation of propane[J]. Microporous and Mesoporous Materials, 2009, 126(1): 152-158.
[34] ZHANG Y, ZHOU Y, QIU A, et al. Propane dehydrogenation on PtSn/ZSM-5 catalyst: Effect of tin as a promoter[J]. Catalysis Communications, 2006, 7(11): 860-866.
[35] 余长林, 葛庆杰, 徐恒泳, 等. 助剂对Pt/γ-Al2O3催化剂丙烷脱氢性能的影响[J]. 石油化工, 2006, 35(3):217-220.(YU Chonglin, GE Qingjie, XU Hengyong, et al. Effect of promoters on dehydrogenation of propane over Pt/γ-Al2O3catalyst[J]. Petrochemical Technology, 2006, 35(3):217-220.)
[36] 杨维慎, 吴荣安, 林励吾. PtSn/Al2O3负载型催化剂丙烷脱氢性能的改进[J]. 石油化工, 1992, 21(8): 511-515.(YANG Weishen, WU Rongan, LIN Liwu. Improvement of catalytic performance of PtSn/Al2O3catalyst for dehydrogenation of propane[J]. Petrochemical Technology, 2006, 35(3):217-220.)
[37] PISDUANGDAW S, PANPRANOT J, CHAISUK C, et al. Flame sprayed tri-metallic Pt-Sn-X/Al2O3catalysts (X= Ce, Zn, and K) for propane dehydration[J]. Catalysis Communications, 2011, 12(12): 1161-1165.
[38] LIU X, FRANK B, ZHANG W, et al. Carbon-catalyzed oxidative dehydrogenation ofn-butane: Selective site formation during sp3-to-sp2 Lattice rearrangement[J]. Angewandte Chemie International Edition, 2011, 50(14): 3318-3322.
[39] SU D S, DELGADO J J, LIU X, et al. Highly ordered mesoporous carbon as catalyst for oxidative dehydrogenation of ethylbenzene to styrene[J]. Chemistry-An Asian Journal, 2009, 4(7): 1108-1113.
[40] LIU L, DENG Q F, AGULA B, et al. Synthesis of ordered mesoporous carbon materials and their catalytic performance in dehydrogenation of propane to propylene[J]. Catalysis Today, 2012, 186(1): 35-41.
[41] MA T Y, LIU L, YUAN Z Y. Direct synthesis of ordered mesoporous carbons[J]. Chemical Society Reviews, 2013, 42(9): 3977-4003.
[42] MICHALAKOS P M, KUNG M C, JAHAN I, et al. Selectivity patterns in alkane oxidation over Mg3(VO4)2-MgO, Mg2V2O7, and (VO)2P2O7[J]. Journal of Catalysis, 1993, 140(1): 226-242.
[43] BETTAHAR M M, COSTENTIN G, SAVARY L, et al. On the partial oxidation of propane and propylene on mixed metal oxide catalysts[J]. Applied Catalysis A: General, 1996, 145(1): 1-48.
[44] BLASCO T, NIETO J M. Oxidative dyhydrogenation of short chain alkanes on supported vanadium oxide catalysts[J]. Applied Catalysis A: General, 1997, 157(1): 117-142.
[45] PANTAZIDIS A, BUCHOLZ S A, ZANTHOFF H W, et al. A TAP reactor investigation of the oxidative dehydrogenation of propane over a V-Mg-O catalyst[J]. Catalysis Today, 1998, 40(2): 207-214.
[46] 方智敏, 翁维正, 万惠霖, 等. VMgO催化剂上丙烷氧化脱氢反应的原位Raman谱学研究[J]. 分子催化, 1998, 12(3):207-213. (FANG Zhimin, WENG Weizheng, WAN Huilin, et al. In situ Raman spectroscopy study on oxidative dehydrogenation of propane over VMgO catalyst[J]. Journal of Molecular Catalysis(China), 1998, 12(3):207-213.)
[47] 刘智勇, 彭志民, 崔湘浩, 等. 钴钼氧化物催化剂对丙烷催化氧化脱氢反应的研究[J]. 分子催化, 1998, 12(3): 193-198. (LIU Zhiyong, PENG Zhiming, CUI Xianghao, et al. Oxidative dehydrogenation of propane over cobalt-molybdenum oxide catalysts[J]. Journal of Molecular Catalysis(China), 1998, 12(3): 193-198.)
[48] 陈树明, 翁维正, 许翩翩, 等. 酸碱性和氧化还原性对负载型钒基催化剂丙烷氧化脱氢性能的影响[J]. 天然气化工, 1998, 3(5):17-20. (CHEN Mingshu, WENG Weizheng, XU Pianpian, et al. Effects of acid-base and redox properties on catalytic performance of supported V-base catalysts for oxidative dehydrogenation of propane[J]. Natural Gas Chemical Industry, 1998, 3(5): 17-20)
[49] 照日格图, 李文钊, 于春英, 等. 钼掺杂LaVO4上丙烷氧化脱氢[J]. 物理化学学报, 2000, 18(1):1-5.(ZHAORI Getu, LI Wenzhao, YU Chunying, et al. Propane oxidative dehydrogenation on molybdenum doped LaVO4catalyst[J]. Acta Phys -Chim Sin, 2000, 18(1):1-5.)
[50] 张昕, 伊晓东, 毕盈, 等. 丙烷选择氧化反应中钼基催化剂动态结构的研究[J]. 催化学报, 2002, 23(2):191-194. (ZHANG Xin, YI Xiaodong, BI Ying, et al. Dynamic structure of Mo-O species in Ag-Mo-P-O catalyst for oxidative dehydrogenation of propane[J]. Chinese Journal of Catalysis, 2002, 23(2):191-194.)
[51] 照日格图, 葛庆杰, 李文钊, 等. Ni-V-O催化剂上丙烷氧化脱氢制丙烯的反应[J]. 催化学报, 2000, 21(4):332-336.(ZHAORI Getu, GE Qingjie, LI Wenzhao, et al. Oxidative dehydrogenation of propane to propylene over Ni-V-O catalysts[J]. Chinese Journal of Catalysis, 2000, 21(4):332-336.)
[52] ZHANG W, ZHOU X, TANG D, et al. Oxidative dehydrogenation of propane over fluorine promoted rare earth-based catalysts[J]. Catalysis Letters, 1994, 23(1-2): 103-106.

[54] GAO X T, RUIZ P, XIN Q, et al. Effect of coexistence of magnesium vanadate phases in the selective oxidation of propane to propene[J]. Journal of Catalysis, 1994, 148(1): 56-67.
[55] CHAAR M A, PATEL D, KUNG H H. Selective oxidative dehydrogenation of propane over VMgO catalysts[J]. Journal of Catalysis, 1988, 109(2): 463-467.
[56] SIEW HEW SAM D, SOENEN V, VOLTA J C. Oxidative dehydrogenation of propane over VMgO catalysts[J]. Journal of Catalysis, 1990, 123(2): 417-435.
[57] 方智敏, 翁维正, 万惠霖, 等. 丙烷氧化脱氢VMgO催化剂活性位的研究[J]. 厦门大学学报(自然科学版), 1998, 37(4):525-531. (FANG Zhimin, WENG Weizheng, WAN Huilin, et al. Study on active sites of VMgO catalysts for oxidative dehydrogenation of propane[J]. Journal of Xiamen University (Natural Science), 1998, 37(4):525-531.)
[58] ROUTRAY K, REDDY K, DEO G. Oxidative dehydrogenation of propane on V2O5/Al2O3and V2O5/TiO2catalysts: Understanding the effect of support by parameter estimation[J]. Applied Catalysis A: General, 2004, 265(1): 103-113.
[59] STELZER J B, CARO J, FAIT M. Oxidative dehydrogenation of propane on TiO2supported antimony oxide/vanadia catalysts[J]. Catalysis Communications, 2005, 6(1): 1-5.
[60] RUSZEL M, GRZYBOWSKA B, GASIOR M, et al. Effect of Au in V2O5/SiO2and MoO3/SiO2catalysts on physicochemical and catalytic properties in oxidation of C3 hydrocarbons and of CO[J]. Catalysis Today, 2005, 99(1-2): 151-159.
[61] PAK C, BELL A T, TILLEY T D. Oxidative dehydrogenation of propane over vanadia-magnesia catalysts prepared by thermolysis of OV (OtBu)3in the presence of nanocrystalline MgO[J]. Journal of Catalysis, 2002, 206(1): 49-59.
[62] KHODAKOV A, OLTHOF B, BELL A T, et al. Structure and catalytic properties of supported vanadium oxides: Support effects on oxidative dehydrogenation reactions[J]. Journal of Catalysis, 1999, 181(2): 205-216.
[64] ZHAORIGETU B, KIEFFER R, HINDERMANN J P. Oxidative dehydrogenation of propane on rare earth vanadates influence of the presence of CO2in the feed[J]. Studies in Surface Science and Catalysis, 1996, 101: 1049-1058.
[65] BATIOT C, HODNETT B K. The role of reactant and product bond energies in determining limitations to selective catalytic oxidations[J]. Applied Catalysis A: General, 1996, 137(1): 179-191.
[66] 方智敏, 翁维正, 万惠霖, 等. 载体酸碱性对钒基催化剂丙烷氧化脱氢性能的影响及其IR和EPR研究[J]. 天然气化工, 1998, 23(2): 25-29. (FANG Zhimin, Weng Weizheng, WAN Huilin, et al. Effects of acidity and basicity of supports on the performance of V-based catalysts for oxidative dehydrogenation of propane and their IR and EPR studies[J]. Natural Gas Chemical Industry, 1998, 23(2): 25-29)
[67] 张伟德, 沙开清, 李基涛, 等. 负载型V2O5/AlPO4催化剂在丙烷氧化脱氢中的催化作用[J]. 天然气化工, 1998, 23(5):21-24. (ZHANG Weide, SHA Kaiqing, LI Jitao, et al. Catalysis of supported V2O5/AlPO4catalysts in oxidative dehydrogenation of propane[J]. Natural Gas Chemical Industry, 1998, 23(5): 21-24)
[68] HABER J, LALIK E. Catalytic properties of MoO3revisited[J]. Catalysis Today, 1997, 33(1): 119-137.
[69] MEUNIER F C, YASMEEN A, ROSS J R H. Oxidative dehydrogenation of propane over molybdenum-containing catalysts[J]. Catalysis Today, 1997, 37(1): 33-42.
[70] LEZLA O, BORDES E, COURTINE P, et al. Synergetic effects in the Ni-Mo-O system: Influence of preparation on catalytic performance in the oxidative dehydrogenation of propane[J]. Journal of Catalysis, 1997, 170(2): 346-356.
[71] MAZZOCCHIA C, ABOUMRAD C, DIAGNE C, et al. On the NiMoO4oxidative dehydrogenation of propane to propene: Some physical correlations with the catalytic activity[J]. Catalysis Letters, 1991, 10(3-4): 181-191.
[72] UEDA W, LEE K H, YOON Y S, et al. Selective oxidative dehydrogenation of propane over surface molybdenum-enriched MgMoO4catalyst[J]. Catalysis Today, 1998, 44(1): 199-203.
[73] 韩智三, 伊晓东, 林洪, 等. Mo-V-Co-O催化剂的丙烷氧化脱氢性能研究[J]. 厦门大学学报(自然科学版), 2005, 44(1):67-70. (HAN Zhisan, YI Xiaodong, LIN Hong, et al. The oxidation dehydrogenation of propane over Mo-V-Co-O catalysts[J]. Journal of Xiamen University (Natural Science), 2005, 44(1):67-70.)
[74] AU C T, ZHANG W D. Oxidative dehydrogenation of propane over rare-earth orthovanadates[J]. J Chem Soc, Faraday Trans, 1997, 93(6): 1195-1204.
[75] FANG Z M, HONG Q, ZHOU Z H, et al. Oxidative dehydrogenation of propane over a series of low-temperature rare earth orthovanadate catalysts prepared by the nitrate method[J]. Catalysis Letters, 1999, 61(1-2): 39-44.
[76] ZHANG W D, AU C T, LI J T, et al. The active sites of the reference phase of SmVO4as catalyst for propane oxidative dehydrogenation[J]. Chemical Research in Chinese Universities, 1998, 14(3): 294-296.
[77] 张伟德, 万惠霖, 蔡启瑞. 纯稀土基丙烷氧化脱氢制丙烯催化剂的研究[J]. 高等学校化学学报, 1993, 14(4): 566-567. (ZHANG Weide, WAN Huilin, CAI Qirui. Studies on propane oxidative dehydrogenation over pure rare earth-based catalysts[J]. Chemical Journal of Chinese Universities, 1993, 14(4): 566-567.)
[78] ZHANG J, SU D, ZHANG A, et al. Nanocarbon as robust catalyst: Mechanistic insight into carbon-mediated catalysis[J]. Angewandte Chemie International Edition, 2007, 119(38): 7460-7464.
[79] FRANK B, ZHANG J, BLUME R, et al. Heteroatoms increase the selectivity in oxidative dehydrogenation reactions on nanocarbons[J]. Angewandte Chemie International Edition, 2009, 48(37): 6913-6917.
[80] QI W, LIU W, ZHANG B, et al. Oxidative dehydrogenation on nanocarbon: Identification and quantification of active sites by chemical titration[J]. Angewandte Chemie International Edition, 2013, 52(52): 14224-14228.
[81] CHEN C, ZHANG J, ZHANG B, et al. Revealing the enhanced catalytic activity of nitrogen-doped carbon nanotubes for oxidative dehydrogenation of propane[J]. Chemical Communications, 2013, 49(74): 8151-8153.
[82] BAEK J, YUN H J, YUN D, et al. Preparation of highly dispersed chromium oxide catalysts supported on mesoporous silica for the oxidative dehydrogenation of propane using CO2: Insight into the nature of catalytically active chromium sites[J]. ACS Catalysis, 2012, 2(9): 1893-1903.
[83] ZHANG F, WU R, YUE Y, et al. Chromium oxide supported on ZSM-5 as a novel efficient catalyst for dehydrogenation of propane with CO2[J]. Microporous and Mesoporous Materials, 2011, 145(1): 194-199.
[84] MICHORCZYK P, PIETRZYK P, OGONOWSKI J. Preparation and characterization of SBA-1-supported chromium oxide catalysts for CO2assisted dehydrogenation of propane[J]. Microporous and Mesoporous Materials, 2012, 161: 56-66.
[85] TAKAHARA I, CHANG W C, MIMURA N, et al. Promoting effects of CO2on dehydrogenation of propane over a SiO2-supported Cr2O3catalyst[J]. Catalysis Today, 1998, 45(1): 55-59.
[86] WU R, XIE P, CHENG Y, et al. Hydrothermally prepared Cr2O3-ZrO2as a novel efficient catalyst for dehydrogenation of propane with CO2[J]. Catalysis Communications, 2013, 39: 20-23.
[87] MICHORCZYK P, OGONOWSKI J. Dehydrogenation of propane to propene over gallium oxide in the presence of CO2[J]. Applied Catalysis A: General, 2003, 251(2): 425-433.
[88] XU B, ZHENG B, HUA W, et al. Support effect in dehydrogenation of propane in the presence of CO2over supported gallium oxide catalysts[J]. Journal of Catalysis, 2006, 239(2): 470-477.
[89] CHEN M, XU J, SU F Z, et al. Dehydrogenation of propane over spinel-type Gallia-alumina solid solution catalysts[J]. Journal of Catalysis, 2008, 256(2): 293-300.
[90] WU J L, CHEN M, LIU Y M, et al. Sucrose-templated mesoporousβ-Ga2O3as a novel efficient catalyst for dehydrogenation of propane in the presence of CO2[J]. Catalysis Communications, 2013, 30: 61-65.
[92] CHEN M, XU J, CAO Y, et al. Dehydrogenation of propane over In2O3-Al2O3mixed oxide in the presence of carbon dioxide[J]. Journal of Catalysis, 2010, 272(1): 101-108.
[93] CHEN M, WU J L, LIU Y M, et al. Study in support effect of In2O3/MOx(M= Al, Si, Zr) catalysts for dehydrogenation of propane in the presence of CO2[J]. Applied Catalysis A: General, 2011, 407(1): 20-28.
[94] TAKEHIRA K, OHISHI Y, SHISHIDO T, et al. Behavior of active sites on Cr-MCM-41 catalysts during the dehydrogenation of propane with CO2[J]. Journal of Catalysis, 2004, 224(2): 404-416.
[95] WECKHUYSEN B M, SCHOONHEYDT R A. Alkane dehydrogenation over supported chromium oxide catalysts[J]. Catalysis Today, 1999, 51(2): 223-232.
[96] NAKAGAWA K, OKAMURA M, IKENAGA N, et al. Dehydrogenation of ethane over gallium oxide in the presence of carbon dioxide[J]. Chemical Communications, 1998 (9): 1025-1026.
[97] MICHORCZYK P, OGONOWSKI J. Dehydrogenation of propane in the presence of carbon dioxide over oxide-based catalysts[J]. Reaction Kinetics and Catalysis Letters, 2003, 78(1): 41-47.
[98] ZHENG B, HUA W, YUE Y, et al. Dehydrogenation of propane to propene over different polymorphs of gallium oxide[J]. Journal of Catalysis, 2005, 232(1): 143-151.
[99] CHEN M, XU J, LIU Y M, et al. Enhanced activity of spinel-type Ga2O3-Al2O3mixed oxide for the dehydrogenation of propane in the presence of CO2[J]. Catalysis Letters, 2008, 124(3-4): 369-375.
Advance in Catalysts for Propane Dehydrogenation to Propylene
ZHANG Lingfeng1, LIU Yalu1, HU Zhongpan1, YANG Yuwang2, YU Haibin2, YUAN Zhongyong1
(1.KeyLaboratoryofAdvancedEnergyMaterialsChemistry(MinistryofEducation),CollaborativeInnovationCenterofChemicalScienceandEngineering(Tianjin),CollegeofChemistry,NankaiUniversity,Tianjin300071,China;2.CNOOCTianjinChemicalResearch&DesignInstitute,Tianjin300131,China)
Propylene is an important chemical in petrochemical industry, and the demand for propylene is continuously increasing. The technologies for dehydrogenation of propane to propylene have
much attention because the conventional petroleum cracking can’t satisfy the growing demand for propylene. Several kinds of the reaction including direct dehydrogenation and oxidative dehydrogenation in the presence of O2or CO2, as well as thermodynamics and mechanism and the corresponding catalysts are reviewed here. Meanwhile, the merits and drawbacks of each catalytic system are discussed, and the further development for the propane dehydrogenation is also presented.
propane;propylene;dehydrogenation;catalyst
2014-11-01 第一作者: 张凌峰,男,博士研究生,从事工业催化的研究
袁忠勇,男,教授,博士,从事纳米催化材料方面的研究;Tel:022-23509610; E-mail:zyyuan@nankai.edu.cn
1001-8719(2015)02-0400-18
O643.36+4
A
10.3969/j.issn.1001-8719.2015.02.019
