脱镁叶绿酸-a甲酯外接环的立体选择性化学反应
2015-06-23张善国李彦龙王进军
张善国,李彦龙,王进军
(烟台大学化学化工学院,山东烟台264005)
脱镁叶绿酸-a甲酯外接环的立体选择性化学反应
张善国,李彦龙,王进军
(烟台大学化学化工学院,山东烟台264005)
以脱镁叶绿酸-a甲酯1为起始原料,利用其的五元外接环的多官能团活性反应区域,分别进行了羟醛缩合反应和还原反应,合成出具有差向异构特征的叶绿素类二氢卟吩衍生物2和3.同时,提出了相应的化学反应机理,并对差向异构体的立体结构进行讨论和表征,解释和总结了脱镁叶绿酸-a甲酯中E-环化学反应的立体选择性.
叶绿素-a;脱镁叶绿酸;化学反应;立体选择性;反应机理
外接五元环系部分是叶绿素-a的标示性结构,其131-位酮羰基和132-位酯羰基组成了典型的β-酮酯活性反应区域,连接于131-位碳上的氢原子一方面可以作为活泼的羰基α-氢,另外与大环色基间的共轭关系使得其质子处于类似于苄基的化学环境中,因而表现出较强的酸性.脱镁叶绿酸是叶绿素-a的最初降解产物,利用外接环上的活性反应区域进行各种结构修饰是获取其他叶绿素降解产物的必经途径,也是合成各种新型叶绿素类二氢卟吩衍生物的重要方法[1-3].因此,利用脱镁叶绿酸外接E-环的特定结构发现和挖掘新的化学反应是叶绿素化学的重要研究内容,在理论研究和实际应用方面均具有非常的意义.
作为天然产物的叶绿素-a的分子中含有3个手性碳原子,所连接的原子或者原子团对叶绿素衍生物的立体化学能够形成程度不同的影响,特别是132-位甲氧甲酰基和17-位的甲氧甲酰乙基距离E-环较近,有时对相应的反应取向甚至起到决定性作用.然而,许多叶绿素衍生物的合成工作都设法回避立体异构体的形成,因此关于脱镁叶绿酸的立体化学研究报告也相对较少,为了深层次地了解叶绿素化学反应和合成具有手性特征的新型多取代二氢卟吩衍生物,本研究在前期工作的基础上[4-9],以脱镁叶绿酸的E-环反应作为基点,合成完成具有差向异构特征的叶绿素类二氢卟吩衍生物的合成.同时,根据相应的化学反应机理和结构表征,对脱镁叶绿酸-a甲酯中E-环化学反应的立体选择性进行了讨论.
在碘素的催化下,脱镁叶绿酸-a甲酯(1,简写为MPa)与多聚甲醛发生了Blanc亲核加成反应,生成一对差向异构体132(S)-132-羟甲基脱镁叶绿酸-a甲酯(2a-b,43%);选用硼氢化钠作为还原剂,在二氯甲烷和甲醇的混合溶液中对起始原料1的外接环的酮羰基实施还原,结果顺利地分离出39%的131(S/R)-131-羟基脱镁叶绿酸-a甲酯(3a-b).为了对照说明脱镁叶绿酸-a甲酯的立体化学特性,脱镁叶绿酸-a甲酯1在乙酸中回流,脱去其132-位酯基而转化成焦脱镁叶绿酸-a甲酯(4,简写为MPPa).合成路线见图1.
1 实验部分
1.1 仪器与试剂
元素分析用Perkin-Elmer 2400型元素分析仪测定;红外光谱用Perkin-Elmer 1730型红外分光光度仪测定(KBr压片法);紫外-可见光光谱用UV-160A型紫外分光光度计测定;核磁共振氢谱用Beucker ARX-300型核磁共振仪测定,内标为TMS.脱镁叶绿酸-a甲酯1按照文献[10]制备.

图1 脱镁叶绿酸的E-环化学反应及其立体选择性Fig.1 The chemical reactions on E-ring of pheophorbide and stereo-selectivities
1.2132(S/R)-132-羟甲基脱镁叶绿酸甲酯(2)的合成
将500 mg MPa 1(0.825 mmol)、250 mg多聚甲醛和150 mg碘素溶解于10 mL二氯甲烷中,室温搅拌20 h,加30 mL水和15 mL二氯甲烷分层萃取,有机相水洗2次,经无水硫酸钠干燥后减压浓缩,硅胶柱层析分离(洗脱剂:V石油醚∶V乙酸乙酯=3∶1),得226 mg墨绿色化合物2a-b(0.355 mmol,43%,a∶b= 20∶19);mp:214~126℃.UV-vis(CH2Cl2)λmax: 413(1.00),505(0.11),535(0.08),610(0.08),667(0.42)nm;1H NMR(CDCl3)δ:-1.80(br s,1H,NH),0.02(br s,1H,NH),1.68(d,J=7.5 Hz,3H,18-CH3),1.71(t,J=7.6 Hz,3H,8-CH3),1.98~2.16,2.20~2.34,2.41~2.61(each m,all 4H,17a+17b-H),3.43,3.56,3.62,3.65,3.72(each s,each 3H,CH3+OCH3),3.71 (q,J=7.6 Hz,2H,8a-H),4.43~4.52(m,1H,18-H),4.68(d,J=8.5 Hz 1H,17-H),4.90 (br s,2H,132-CH2),5.52(br s,132-OH),6.21(d,J=11.6 Hz,cis-3b-H),6.31(d,J= 17.8 Hz,1H,trans-3b-H),8.04(dd,J=17.8,11.6 Hz,1H,3a-H),9.63,9.49,8.63(each s,each 1H,meso-H).2b:δ:-1.72(br s,1H,NH), 0.02(br s,1H,NH),1.60(d,J=7.5 Hz,3H,18 -CH3),1.70(t,J=7.6 Hz,3H,8-CH3),1.98~2.16,2.20~2.34,2.41~2.61(each m,all 4H,17a+17b-H),3.26,3.42,3.62,3.64,3.70 (each s,each 3H,CH3+OCH3),3.71(q,J=7.6 Hz,2H,8a-H),4.43~4.52(m,1H,18-H),4.15(d,J=9.0 Hz,1H,17-H),4.90(br s,2H,132-CH2),5.33(br s,132-OH),6.21(d,J= 11.6,1H,cis-3b-H),6.30(d,J=17.8 Hz,1H,trans-3b-H),8.00(dd,J=17.8,11.6 Hz,1H,3a-H),8.61,9.46,9.60(each s,each 1H,meso -H).IR(KBr)ν:3 446(N—H),2 963(C—H),1 739~1 689(==CO),1 672(==CC),1 541(chlorin skeleton),1 527,1 458,1 398,1 170,1 068,987 cm-1;EI-MS m/z:637.3(MH+);Anal. calcd for C37H40N4O6:C 69.79,H 6.33,N 8.80; found C 69.69,H 6.10,N 8.98.
1.3 132(S/R)-131-羟基-131-去氧脱镁叶绿酸甲酯(3)的合成
将280 mg MPa 1(0.462 mmol)溶于10 mL二氯甲烷中,然后加入15 mL甲醇稀释,在0℃条件下,缓慢滴加溶解于5 mL甲醇的93 mg硼氢化钠,室温避光搅拌反应24 h,加入20 mL二氯甲烷和30mL水分层,萃取,有机相水洗,无水硫酸钠干燥,减压蒸除溶剂,硅胶柱层析分离(洗脱剂:V石油醚∶V乙酸乙酯=2∶1),得112 mg墨绿色固体3a(0.061 mmol),产率26%和3b(0.031 mmol),产率13%. 3a:UV-vis(CH2Cl2)λmax:397(1.00),499 (0.09),532(0.06),607(0.05),653(0.26) nm;1H NMR(CDCl3)δ:-3.24(br s,1H,NH),-1.27(br s,1H,NH),1.74(t,J=7.6 Hz,3H,8-CH3),1.85(d,J=7.2 Hz,3H,18-CH3),2.02~2.26,2.23~2.32,2.38~2.56,2.58~2.68 (each m,all 4H,17a+17b-H),3.80(q,J=7.6 Hz,2H,8a-H),3.36,3.50,3.53,3.60,3.89 (each s,each 3H,OCH3+CH3),4.39(dd,J= 8.5,2.1 Hz,1H,17-H),4.58(qd,J=7.4,1.4 Hz,1H,18-H),6.14(d,J=11.5 Hz,1H,cis-3b-H),6.31(d,J=17.8 Hz,1H,trans-3b-H),6.34(d,J=6.7 Hz,1H,132-H),6.71(d,J =6.7 Hz,1H,131-H),8.17(dd,J=17.8,11.5 Hz,1H,3a-H),9.85,9.65,8.92(each s,each 1H,meso-H).3b:1H NMR(CDCl3)δ:-3.18(br s,1H,NH),-1.29(br s,1H,NH),1.73(t,J= 7.6 Hz,3H,8-CH3),1.88(d,J=7.2 Hz,3H,18-CH3),2.02~2.26,2.23~2.32,2.38~2.56,2.58~2.68(each m,all 4H,17a+17b-H),3.78 (q,J=7.6 Hz,2H,8a-H),3.10,3.37,3.53,3.58,3.82(each s,each 3H,OCH3+CH3),4.50 (dd,J=8.5,2.1 Hz,1H,17-H),4.55(qd,J= 7.3,1.4 Hz,1H,18-H),6.13(d,J=11.5 Hz,1H,cis-3b-H),6.31(d,J=17.8 Hz,1H,trans -3b-H),6.33(s,1H,132-H),6.61(br s,1H,131-H),8.16(dd,J=17.8,11.5 Hz,1H,3a-H),8.91,9.62,9.83(each s,each 1H,meso-H).IR(KBr)ν:3 434(N—H),2 968(C—H),1 741~1 701(==CO),1 663(==CC),1 527 (chlorin skeleton),1 521,1 438,1 368,1 174,1 068 cm-1;EI-MS m/z:609.3(MH+);Anal. calcd for C36H40N4O5:C 71.03,H 6.22,N 9.20; found C 71.19,H 6.10,N 9.38.
1.4 焦脱镁叶绿酸-a甲酯(4)的合成
将1 g脱镁叶绿酸-a甲酯1(1.649 mmol)用100 mL冰乙酸溶解后,氮气保护,搅拌回流反应6 h,反应结束后减压蒸馏,蒸除大部分乙酸后加入100 mL水,用二氯甲烷(3×25 mL)萃取,有机相干燥后减压浓缩,将所得浓缩物通过硅胶柱层析分离(洗脱剂:V石油醚∶V乙酸乙酯=3∶1),得到780 mg墨绿色固体2(1.423 mmol),产率为86%.mp:189~192℃,UV-vis(CHCl3)λmax:346(1.45),410 (1.00),508(0.02),536(0.03),610(0.03),670 (0.38)nm;1H NMR(CDCl3)δ:-1.67(br s,1H,NH),0.48(br s,1H,NH),1.70(d,J=7.4 Hz,3H,18-CH3),1.81(t,J=7.5 Hz,3H,8b-CH3),2.15~2.37,2.40~2.80(each m,all 4H,17a+17b-H),3.25,3.41,3.68,3.61(each s,each 3H,OCH3+CH3),3.70(q,J=7.2 Hz,2H,8a-H),4.18~4.35(m,1H,17-H)4.38~4.52 (m,1H,18-H),5.11(d,J=20.0 Hz,1H,132-H),5.27(d,J=20.0 Hz,1H,132-H),6.18~6.29(m,2H,3b-H),8.02(m,1H,3a-H),8.56,9.40,9.52(each s,1H,meso-H).其他的分析数据与文献[11]一致.
2 结果与讨论
2.1 (焦)脱镁叶绿酸-a甲酯的立体选择性化学反应
脱镁叶绿酸-a甲酯1的外接E-环中的132-亚甲基受到邻位2个羰基的影响而具有很强的化学反应性,除了132-位碳原子以外,其他原子全部以SP2杂化的形式存在,因而具有非常强直的刚性.在碘催化与多聚甲醛的Blanc反应中,首先是碘素裂分成碘负离子促成多聚甲醛的分解,所生成的次碘酸和碘化氢再相互作用回归成碘和生成一分子水;脱镁叶绿酸的外接E-环羰基可以转换成烯醇式1a,然后烯醇碳碳双键可以在二氢卟吩大环的上下两面与甲醛发生亲核加成反应,进而得到差向异构体132(S)-132-羟甲基脱镁叶绿酸甲酯2a及其异构体2b.当烯醇式双键以a-途径进攻甲醛的时候,亲核加成受到来自17-位长链酯基的空间位阻;如果从相反的方向以b-途径进行相同的反应,17-位的立体效应基本影响不到E-环烯醇对甲醛的亲核进攻.由于17-位酯基距离132-位相对较远,其空间效应的影响力也非常有限,其差向异构体比例仅为20∶19.
当MPa与硼氢化钠进行还原反应时,可以提供氢负离子的复盐还原剂同样有2个取向进攻131-位环酮羰基(图2中A框).从132-甲氧甲酰基取向的反方向进入的氢负离子给出主要产物131(S) -131-羟基脱镁叶绿酸-a甲酯3a,选择与酯基相同方向的还原反应则生成次要产物131(R)-131-羟基脱镁叶绿酸-a甲酯3b.硼氢化钠在还原过程中可以选择2个方向对E-环羰基实施进攻,沿着a-途径的还原反应可以避开邻位甲氧甲酰基的立体排斥,而b-途径的相同反应则遭遇到较大的立体障碍.由于甲氧甲酰基毗邻于E-环羰基,其空间效应对化学反应的立体选择起到非常重要的作用,所以后者的产率不足前者的二分之一(图2). 2
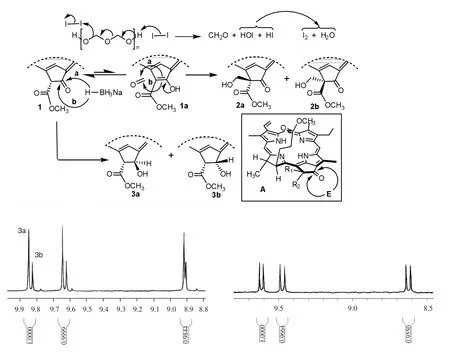
图2 (焦)脱镁叶绿酸-a甲酯外接E-环的立体选择性反应Fig.2 The steroselectivites reactions on the exocyclic E-ring of methyl(pyro)pheophorbide-a
.2差向异构体的核磁共振氢谱与其立体化学结构
脱镁叶绿酸-a甲酯1与甲醛的2个差向异构产物的132-位取代基和17-位质子的化学位移表现出明显的差异,由于处于同侧酯羰基的各向异性的屏蔽作用,使得具有R-构型2a的17-位质子的吸收信号低场移动至4.68处,2b中相对应的吸收峰则出现在4.15的位置上.2个异构体的132-位羟甲基与偕位酯羰基的距离基本一致,因而二者碳上质子的振动频率均在4.90处反映出一个单宽吸收峰.然而,羟甲基中羟基氢的化学位移却相差很大,分别为5.52和5.33.由于脱镁叶绿酸外接E-环的特定立体结构,处于132-位上不同取向的羟基与环上羰基形成氢键的能力不同,所以导致质子的吸收信号形成明显的区别.外接E-环羰基的伸展方向并不是完全处于色基的平面上,实际上是向17-位尾端酯基的伸展方向偏移,所以,对周围氢质子所施加的电子效应则有所不同,例如,MPPa 4在132-位上2个质子的氢谱吸收分别为5.27和5.11,化学位移处于相对低场的质子与132-位碳所形成的化学键的伸展方向指向大环色基的上方,与E-环羰基相互平行,其碳氧双键的去屏蔽作用导致2个偕碳质子的核磁振动频率产生差异.2a的132-位羟基空间取向与环羰基同向,因而可以形成分子内氢键,而2b中处于相反方向的羟基则距离环羰基太远,所以不能与其建立任何联系.当氢键形成时,所产生的静电场将氢原子拉向羰基,同时将羟基氢氧键的电子推向氧原子,结果使得氢原子周围的电子云密度降低,因而去屏蔽效应增加,其质子的化学位移也向低场推移.
差向异构体3a和3b E-环上的氢质子所表现的吸收峰截然不同,3b中处于同一方向的2个质子之间的二面角相对较小,因此二者之间没有形成偶合,其吸收信号均为一单峰;而3a中处于相反方向的2个质子之间相应夹角比较大,其吸收峰裂分为双重峰,二者之间的偶合常数为6.7 Hz.
与异构体2a-b一样,3b中处于同一方向的邻位羟基和酯羰基可以形成氢键,而3a的伸向2个方向的含氧取代基团则很难相互作用.氢键的建立使得3b羟基氧的周围具有更多的电子云密度,因而其吸电子能力相对降低,所以对应的131-位质子的化学位移出现在相对低场;3a的131-位氢原子与环上酯基处于同侧,碳氧双键的各向异性促成其吸收信号低场移动.差向异构体3中132-位质子所邻的立体环境大致相同,因而其化学位移也基本一致(Δδ= 0.01).3a和3b在7位上的单氢化学位移也表现出比较大的差异,其Δδ达到0.21,其原因主要是由于3b的酯羰基受到氢键的束缚而具有一定的刚性,而异构体3a的酯羰基则能够自由旋转,因此,前者对17 -位质子的去屏蔽力度要高于后者(图3).

图3 叶绿素差向异构体的核磁共振氢谱与其立体化学结构Fig.3 The1H NMR spectra of chlorophllous epimers and their stereochemical structures
[1]王进军.叶绿素-a衍生物的化学反应和多取代卟吩(啉)合成的研究进展[J].有机化学,2005,11(25):1153 -1371.
[2]Chen Yihui,Lin Guolin,Pendey R K.Synthesis of bacteriochlorind and their potential untility in photodynamic[J]. Current Org Chem,2004,8(12):1105-1134.
[3]Pavlov V Y,Ponomarev G V.Modification of the peripheral substituents in chlorophylls a and b and their derivatives[J].Chem Heterocyclic Comd,2004,40(4):393-425.
[4]徐希森,刘洋,张善国,等.132-氧代脱镁叶绿酸-a甲酯的E-环反应及其最大可见光吸收波长的变化[J].烟台大学学报:自然科学与工程版,2013,26(4):29-34.
[5]王振,杨泽,刘洋,等.焦脱镁叶绿酸-a甲酯外接环的氧化和还原反应[J].烟台大学学报:自然科学工程版,2013,26(1):34-39.
[6]于沙沙,徐希森,刘洋,等.周环上芳基取代的叶绿素类二氢卟吩衍生物的合成[J].有机化学,2014,34 (002):362-370.
[7]刘洋,徐希森,李家柱,等.叶绿素降解产物的环丙基化及其二氢卟吩衍生物的合成[J].有机化学,2014,34 (3):552-560.
[8]王振,杨泽,刘洋,等.焦脱镁叶绿酸-d甲酯的化学修饰与叶绿素类二氢卟吩衍生物的合成[J].有机化学,2012,32(12):2300-2308.
[9]杨泽,王振,刘洋,等.具有长波吸收的(焦)脱镁叶绿酸衍生物的合成及其电子光谱[J].有机化学,2013,33 (01):116-124.
[10]Smith K M,Bissert G M F,Bushell M J.Partial synthesis of optically pure methyl bacateriophophorbide-a and d from methyl pheophorbide a[J].J Org Chem,1980,45(11): 2218-2224.
[11]李家柱.具有叶绿素碳架的光动力抗癌药物的合成研究[D].烟台:烟台大学,2008.
Stereoselectivitic Chemical Reactions of Methyl Pheophorbide-a ZHANG Shan-guo,LI Yan-long,WANG Jin-jun
(School of Chemistry and Chemical Engineering,Yantai University,Yantai 264005,China)
Methyl pheophorbide-a 1 is used as starting material.The Aldol reaction and reduction are performed,respectively,by using multi-functional active region on the five-membered exocyclic ring to synthesize chlorophyllous chlorins 2 and 3 with stereo-isomeric feature.The corresponding reaction mechanisms are proposed,and the stereo-structures of epimers are discussed and characterized.The stereoselectivity of chemical reactions on the E-ring of methyl pheophorbide-a is explained and summarized.
chlorophyll-a;pheophorbide;chemical reaction;stereo selectivity;reaction mechanism
O644.1
A
(责任编辑 周雪莹)
1004-8820(2015)02-0084-06
10.13951/j.cnki.37-1213/n.2015.02.002
2014-04-06
国家自然科学基金资助项目(21272048);国家科技部中匈政府间合作项目(2013年).
张善国(1988-),男,山东德州人,硕士研究生.
王进军(wjj1955@163.com),教授,博士,研究方向:有机合成.
