乙炔加氢反应系统操作优化策略
2015-06-19田亮蒋达钱锋
田亮,蒋达,钱锋
(华东理工大学化工过程先进控制与优化技术教育部重点实验室,上海 200237)
引 言
高温裂解石脑油或 C2~C6饱和烃类制备聚合级乙烯是石油化工的一个重要工业过程[1-2],裂解产物包含大量乙烯和少量的乙炔(体积分数 0.5%~2%)。因为乙炔会使下游用于进行乙烯聚合的催化剂中毒[3],它的含量必须被降低到10−6级。现代石油化工可以采用负载的钯金属催化剂,通过乙炔选择性催化加氢有效地从富乙烯流股中除去乙炔,同时没有大量乙烯加氢。对于富乙烯流中的乙炔的加氢过程,工业上有两种基本方法可供选择,分别称为后加氢和前加氢过程[1-2]。这两种方法的不同之处在于乙炔加氢反应器在整个乙烯装置流程中所处的位置,后加氢法的反应器位于脱乙烷单元之后,所以入口流股主要包含碳二烃类。
多年来研究人员对碳二馏分选择加氢催化剂的研究从来没有中断过,在开发低成本、高活性、高乙烯选择性、高空速和低绿油生成量的碳二选择加氢催化剂方面已取得了一定进展,从载体和添加助催化剂组分着手改进催化剂的性能,无须加入一氧化碳来调节乙烯选择性的加氢催化剂是近年来该领域最为突出的一个研究成果。国内的催化剂牌号有北京化工研究院的BC和兰州化工研究院的LY。
乙炔和乙烯加氢是中度的放热反应,在后加氢反应器中,入口物料较低的含氢量容易导致含碳组分在催化剂上的沉积,从而引起催化剂钝化速度加快。为了保证出口物料中对乙炔的含量要求,需要通过增加反应的操作温度来进行补偿钝化作用。一般每经过几个月的操作,就要进行一次催化剂再生。活性和选择性通过从进料到催化剂床层的温度和氢炔比等因素进行控制[4]。已有工作研究了乙炔加氢反应器的控制系统,投入使用后稳定了加氢反应过程[5-7],但工厂实际使用过程中常注重单个反应器的运行状况,而没有考虑反应器组合的性能,即在反应系统中,各个反应器中催化剂的性能是不一致的,所承担的除炔负荷需要优化配置。
本文涉及的乙炔加氢反应系统由两段反应器串联构成,一段反应器转化大部分乙炔、二段反应器转化剩余的乙炔,使得乙炔含量减少到 1×10−6以下。在保证产品合格条件下,尽量减少氢气使用量,使乙炔最大程度地转化为乙烯。乙炔加氢过程中,催化剂使用时间、反应温度、氢炔比、一段和二段的除炔比例都将影响最终的乙烯增量[8-9]。
催化剂失活是与温度密切相关的,温度越高,越容易失活,从催化剂失活的角度看温度越低越好。然而如果低于触发反应的临界温度点,反应器出口的乙炔浓度就会波动剧烈从而影响产品质量[10]。早期已确定的许多重要的特性之一就是钯金属可以让乙炔选择性地加氢生成乙烯。在一个恒容系统中,只要有乙炔存留,即使有多余的氢存在,也可以维持很高的选择性,然而这要求系统温度在室温范围内,高温则会促使生成乙烷的反应的发生[11]。
综合以上两个方面,在保证催化剂活性的条件下尽量维持稳定的入口温度,一段反应器设定为38℃,而二段反应器入口温度设定为45℃,入口温度不作为提高乙烯增量的决策变量。
实际生产过程中没有数学模型支撑,各反应器的除炔负荷分配按照经验值来设定,常常出现不合理的操作状况[12-13]。例如,当发现产品不合格时,工业上的操作主要是加氢使得产品不再“漏炔”,当加入过量的氢气仍然无法转化乙炔时,才考虑切换反应器对催化剂再生,显然这种操作在催化剂使用后期大大降低了加氢选择性。因此本文通过已建立的加氢过程模型,采用优化方法寻求最佳氢炔比,可以在保证产品质量的前提下,提高乙烯产量。增加效益主要考虑以下两个方面:① 合理分配各段反应器的除炔负荷;② 合理设计催化剂切换再生策略。
1 碳二加氢反应过程模型
1.1 选择催化加氢原理
准确的动力学是成功模拟反应器的性能的基础,可以用来寻找合适的温度和压力从而提高乙炔加氢生成乙烯的选择性,同时减少氢气使用量。在评价了十多种乙炔选择加氢动力学模型基础上,挑选出一种最符合实际工况的动力学方程组用于模拟反应过程[14]。反应动力学方程式如下
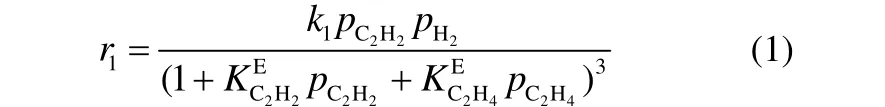

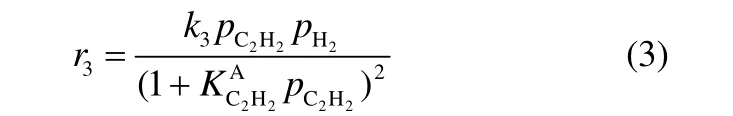
该模型所表示的加氢反应机理主要由波兰科学院Borodzinski教授提出[12]。在原文献中存在乙炔加氢生成乙烷的反应,但由于发现该反应速率较小,故不考虑这个反应。Borodzinski[15-16]首次应用该模型来描述碳沉积物对乙炔-乙烯混合物在钯催化剂上加氢过程的影响。通过碳氢化合物覆盖层在反应过程中的沉积,在钯金属表面会形成两种不同的催化中心:A和E中心,分别进行乙炔和乙烯的转化。A中心由一系列狭窄的吸附中心构成,它们可以吸附乙炔和氢气,但是因为太狭窄而不能吸附乙烯,因而不能进行乙烯加氢;E中心可以吸附包括乙烯在内的所有反应物,由于稳态催化剂上E中心的数量要远远小于A中心的数量,E中心上的乙炔加氢速率可以忽略不计,主要对乙烯进行加氢反应。在E中心上,乙烯加氢生成乙烷的反应是通过朗格缪尔-欣谢尔伍德机理进行的[式(1)]。A中心上的乙炔选择性加氢生成乙烯主要通过两个平行反应进行:① 乙炔与从碳沉积物上转移的氢发生反应[式(2)];②乙炔与竞争性吸附的氢发生反应[式(3)]。
1.2 反应器模型
本文采用等压、绝热、拟均相、一维平推流模型用于模拟加氢反应过程。

其中,Fi是气体i的摩尔流速,kmol·h−1;ρB是催化剂密度,kg·m−3;Cp,i是气体i比定压热容,kJ·kg−1·K−1;rj是反应j速率,kmol·kg−1·K−1;ΔH是反应焓变,kJ·kmol−1;S是反应器截面积,m2;T是温度,K;z是反应器长度,m。式(4)表示了物料平衡,式(5)表示了能量平衡。
1.3 失活动力学
在催化剂上吸附的乙炔会经历一个缓慢的加氢-低聚反应,生成包含偶数个碳原子的高分子量低聚物。这些低聚物会堵塞催化剂微孔,使得催化剂比表面积下降,从而引起活性下降[17]。催化剂失活是一个复杂的现象,催化剂失活机理和方式取决于各个具体的实际过程[18-19]。对于一个表观速率来说,其表现形式可用式(6)表示

其中ri,d(t,T)是指某一时刻反应物在催化剂上的反应速率,r0i(T)是指反应物在新鲜催化剂上的反应速率。ai(t,T)是指某一时间、一定温度下的失活系数,其表达式如式(7)所示。

2 优化结果及讨论
采集工业数据,使用遗传算法估计出反应动力学和失活动力学参数[20]。对于串联的反应器组合,不同反应器中的乙炔转化分布是一个决策变量,分配的好坏决定着最终的乙烯增量。串联的两组反应器的作用不尽相同,一段反应器转化大部分的乙炔,而二段反应器作为保护段,转化剩余的乙炔,并把它降低到10−6级别。因此这两段的氢炔比是不同的,正常来说二段反应器的氢炔比大于一段反应器,二段反应器常常是过加氢的,以防止出现漏炔现象。在动力学和失活模型已知的情况下[8],设定经过串联的两段反应后乙烯增量最大化为目标函数,并增加产品质量控制约束,即二段反应器出口乙炔和氢气都小于1×10−6,使用编写程序寻优,调整两段反应器的除炔负荷,提高乙烯增量。
图1 显示优化后的两组反应器中氢炔比变化趋势。理想状态下一段反应器几乎不要增加氢炔比,维持在0.9附近,而二段反应器为了实现合格产品氢炔比需要从1.9逐渐增加到3.5,因此总氢炔比一直在上升,从1.4上升到2.0。

图1 最佳的氢炔比变化趋势Fig. 1 Trend of optimal H2/C2H2
在本文中,选择性特指在一定氢气条件下,乙烯增加与乙炔转化量的比值。对于C2加氢反应,在本征选择性不变前提下,氢炔比越小,反应后所呈现的选择性越高。图2显示了相对应的反应器中选择性变化趋势,一段反应器承担大部分的除炔任务,维持较高的选择性(即较低的氢炔比)有利于把乙炔转化成乙烯。尽管二段反应器的选择性下降很多,但是其承担的除炔负荷不高,相应的乙烯的损失量也不会太大。
图3给出了反应器中乙烯增量的变化趋势。一段反应器中乙烯增量从初期的 0.75%降到后期的0.55%左右,而二段反应器从最初的0.10%降到后期的−0.75%。整个反应系统中的乙烯增量之和从最初的0.80%降到后期的−0.20%左右。当氢气加入量过多,副反应加剧,使得过多的乙烯和氢气反应生成乙烷,导致产品收益下降。在实际操作过程中,由于没有合理分配两段反应器的除炔负荷,在后期乙烯损失是很大的。

图2 最佳的选择性变化趋势Fig. 2 Trend of optimal selectivity

图3 最佳的乙烯增量变化趋势Fig. 3 Trend of optimal ethylene increment
在操作条件优化的基础上,通过调整切换周期可以增加乙烯增量。为了计算方便,定义了平均每天乙烯增量,即计算调整切换策略后获得总的乙烯增量除以评估时期。图4给出了催化剂单次使用周期与平均每天乙烯增量的关系。发现一段反应器的使用周期长短对乙烯增量影响不大。如果一段反应催化剂在540 d中没有切换下来再生,二段反应器中催化剂的单次使用周期从540 d降到60 d时,乙烯增量可以从0提高到0.45%。但是较短的单次使用周期将大大增加催化剂再生次数,在这个过程中二段反应器的催化剂再生次数从1次提高到9次,每次再生催化剂也是一笔不小的费用。因此需要权衡再生操作费用和提高总选择性后增加的效益,使之获得最大化效益,式(8)给出了盈利计算公式。表 1给出了相关的费用,每小时原料气处理量为65000 m3,催化剂单次切换再生费用为350000元,设定如下目标函数

图4 切换周期与每日乙烯平均增量的关系Fig. 4 Daily ethylene increment under different regeneration cycles for two reactors

其中,ΔIC2H4是乙烯平均每日增量,%;MC2H4,MC2H6和MH2是乙烯、乙烷和氢气的平均每日质量流量;PC2H4,PC2H6和PH2是乙烯、乙烷和氢气的价格;Preg是催化剂再生费用;tfirst和tsecond分别是一段和二段反应器单次运行天数。

表1 各产品价格Table 1 Products prices
通过优化该目标,可以估算出一个合理的再生策略。图5给出了两组反应器切换周期对整体经济效益增量的影响,该图给出了各种切换再生策略:当两组反应器都使用540 d时,由于其选择性下降严重,整体效益并不好,如果平摊再生催化剂的操作费用,每天需要1296元;当两组反应器都只使用90 d时,尽管其选择性较高,但频繁切换反应器和再生催化剂,其操作费用较高,整体效益较差;其中最差的一种情况是频繁再生一段催化剂的同时单次最长时间使用二段反应器;当一段反应器使用420 d,二段反应器使用120 d,整体的经济效益最大。在负荷分配优化的条件下得到最优的切换周期,扣除再生催化剂的费用,平均每日增加 2185元收益。显然优化切换策略后,增加的收益可达到 127万元/年。
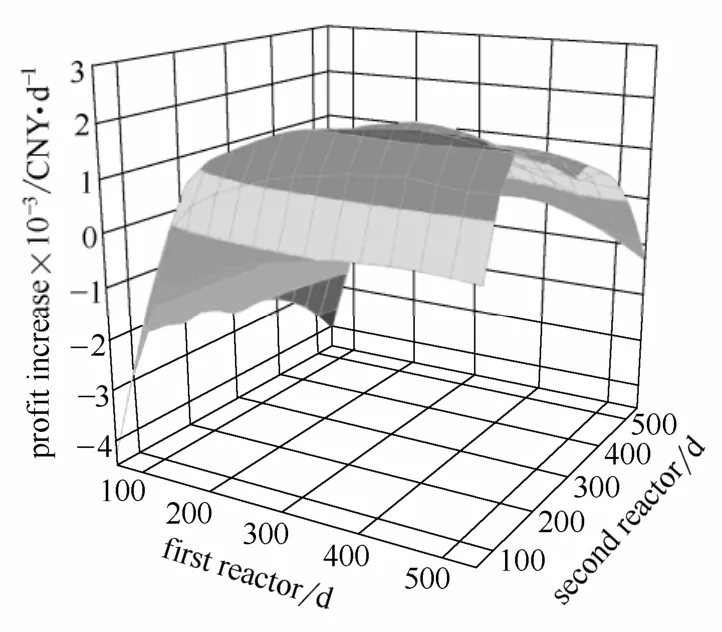
图5 反应器单次使用周期与效益增加的关系Fig. 5 Daily profit increment under different regeneration cycles for two reactors
二段反应器使用4个月后,对于转化微量乙炔反应出现困难,需要更大的氢炔比才能使得乙炔含量降低到10−6级别。但如果切换后直接作为一段反应器使用,此时把乙炔从约1.2%降低到约0.2%时,所需配氢量并不太大,其选择性仍比较高。如果认为在实际运行过程中一段反应器和二段反应器的本征失活趋势相同,那么当二段反应器使用了4个月后,不需再生催化剂而直接切换作为一段反应器使用,这样可以减少一段反应器的再生费用。通过循环操作,一段反应器中的催化剂活性一直保持在4~8月之间,会比单次使用14个月的催化剂的平均选择性要高。
3 结 论
通过数学模型计算,可以合理分配各反应器的乙炔转化率任务。一段反应器的氢炔比尽量不要超过1.0,二段反应器中的氢炔比随着催化剂失活逐渐增加。通过对催化剂再生周期的研究,发现两组催化剂的再生周期不应该相同,一段反应器频繁切换再生并不会提升整体效益,甚至反而会减少整体效益,合理的使用周期是14个月左右。二段反应器的任务是把乙炔含量降到10−6级别,因此要保持较高的选择性,延长单次使用周期会大大降低整体效益,但考虑到再生费用和产品价格,其最佳的切换周期是4个月左右。
[1]Weirauch W. Naphtha, gasoline demand is forecast [J].Hydrocarbon Process, 1998, 77 (9): 25-27
[2]Zehnder S. What are Western Europe’s petrochemical feedstock options? [J].Hydrocarbon Process, 1998, 77 (2): 59-60
[3]Molnár Á, Sárkány A, Varga M. Hydrogenation of carbon–carbon multiple bonds: chemo- regio- and stereo-selectivity [J].J. Molec.Catal. A-Chem., 2001, 173 (1/2): 185-221
[4]Näsi N, Alikoski M, White D C. Advanced control of acetylene reactor [J].Hydrocarbon Process, 1985(6): 57-60
[5]Schbib N S, García M A, Gigola C E, Errazu A F. Kinetics of front-end acetylene hydrogenation in ethylene production [J].Ind.Eng. Chem. Res.,1996, 35(5):1496-1505
[6]Gobbo R, Soares R P, Lansarin M A, Secchi A R, Ferreira J M P.Modeling, simulation, and optimization of a front-end system for acetylene hydrogenation reactors [J].Braz. J. Chem. Eng.,2004,21(4):545-556
[7]Abilov A, Kocak M C. An optimal control application to an industrial hydrogenation reactor [J].Chem. Eng. Res. Des.,2000, 78(4): 630-632
[8]Tian Liang(田亮), Jiang Da(蒋达), Qian Feng(钱锋). Simulation and optimization of acetylene converter with decreasing catalyst activity[J].CIESCJournal(化工学报), 2012, 63(1):185-192
[9]Luo Xionglin(罗雄麟), Liu Jianxin(刘建新), Xu Feng(许锋), Zuo Xin(左信). Heterogeneous two-dimensional dynamic modeling and analysis of acetylene hydrogenation reactor [J].Journal of Chemical Industry and Engineering(China)(化工学报), 2008, 59(6):1454-1461
[10]Asplund S. Coke formation and its effect on internal mass transfer and selectivity in Pd-catalysed acetylene hydrogenation [J].J. Catal.,1996, 158 (1): 267-278
[11]Khan N A, Shaikhutdinov S, Freund H J. Acetylene and ethylene hydrogenation on alumina supported Pd-Ag model catalysts [J].Catal.Lett., 2006,108:159-164
[12]Borodzinski A, Cybulski A. The kinetic model of hydrogenation of acetylene-ethylene mixtures over palladium surface covered by carbonaceous deposits [J].Appl. Catal. A,2000, 198:51-66
[13]Wang Fei(王飞),Luo Na(罗娜),Jiang Da(蒋达),Qian Feng(钱锋).Dynamic simulation and control of acetylene hydrogenation reactor in ethylene plant [J].Computers and Applied Chemistry(计算机与应用化学), 2012, 29(1):85-89
[14]Tian Liang(田亮),Jiang Da(蒋达), Qian Feng(钱锋). Reaction kinetic comparisons for industrial selective hydrogenation of acetylene on palladium catalysts [J].Computers and Applied Chemistry(计算机与应用化学), 2012, 29(9): 1031-1035
[15]Borodzinski A. Hydrogenation of acetylene-ethylene mixtures on commercial palladium catalyst [J].Catal. Lett.,1999, 63 (1-2):35-42
[16]Borodzinski A. Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts(Part 1): Effect of changes to the catalyst during reaction [J].Cat. Rev. - Sci. Eng.,2006, 48: 91-144
[17]Houzvicka J, Pestman R, Ponec V. The role of carbonaceous deposits and support impurities in selective hydrogenation of ethyne [J].Catal.Lett.,1995, 30(1–4):289-296
[18]Webb G. Formation and role of carbonaceous residues in metal-catalyzed reactions of hydrocarbons [J].Catal. Today,1990, 7(2): 139-155
[19]Albers P, Pietsch J, Parker S F. Poisoning ad deactivation of palladium catalysts [J].J. Molec. Catal. A:Chem.,2001, 173 (1–2):275-286
[20]Elliott L, Ingham D B, Kyne A G, Mera N S, Pourkashanian M,Wilson C W. Genetic algorithms for optimisation of chemical kinetics reaction mechanisms [J].Prog. Energy Combust. Sci.,2004, 30:297-328
