有机催化卤代亲核试剂参与的串联反应研究进展
2015-06-07罗年华钟瑜红蔡久彪
罗年华,钟瑜红,汪 波,王 勇,蔡久彪
(1.上饶师范学院,化学化工学院,江西上饶334001;2. 新建三中,江西南昌330100 )
有机催化卤代亲核试剂参与的串联反应研究进展
罗年华1,钟瑜红1,汪 波1,王 勇1,蔡久彪2
(1.上饶师范学院,化学化工学院,江西上饶334001;2. 新建三中,江西南昌330100 )
近年来,有机小分子催化卤代亲核试剂参与的串联反应引起了化学家们的高度重视,他们发展了大量有价值的新型串联反应,制备了许多有用的手性和药物分子。常见的卤代亲核试剂主要包括α或γ-卤代亲核试剂。由于这些卤代亲核试剂的α或γ-位含有卤素,它们与亲电试剂发生第一步的亲核反应后,能够再与亲核试剂发生反应,从而实现两步反应的串联。对近年来有机小分子催化的α或γ-卤代亲核试剂参与的串联反应进行了综述。
亲核试剂;串联反应;卤代;有机催化
串联反应(tandem reaction, cascade reaction, domino reaction) 是指两步或者两步以上的反应在相同的条件下连续进行,而不需要对中间产物进行分离和纯化,一次性得到最终产物的一类反应。串联反应在一些复杂分子的合成中具有非常广泛的应用。有机小分子催化非常适合用于构建串联反应。首先,由于有机催化反应一般在很温和的条件下进行,因此多数有机官能团能够耐受。其次,有机小分子催化反应是多种催化模式共存的,因此容易实现多种反应的串联[1]。近年来,有机小分子催化的串联反应受到了化学家们的高度重视,发展了大量有价值的新型串联反应,制备了许多有用的手性分子。
碳亲核试剂是有机合成中应用最广泛的亲核试剂,一般含有比较活泼的亚甲基或次亚甲基。最常见的碳亲核试剂包括:1,3-二羰基化合物、硝基烷烃、醛酮类以及烯醇硅醚类化合物。如果这些碳亲核试剂的α或γ-位含有卤素,它们与亲电试剂发生第一步的亲核反应后,能够再与亲核试剂发生反应,从而实现两步反应的串联。本文将对近年来有机小分子催化的α或γ-卤代亲核试剂参与的串联反应进行总结和论述。
1 有机催化α-卤代亲核试剂的串联反应
1.1 有机催化α-卤代1,3-二羰基化合物的串联反应
α-卤代丙二酸酯是α-卤代1,3-二羰基化合物中最具有代表性的一类化合物。2007年,Cordova小组报道了α,β-不饱和醛与α-溴代丙二酸酯的不对称串联反应。该反应在手性仲胺二芳基脯氨醇硅醚(3和4)的催化下,得到了非常有用的2-醛基环丙烷类化合物(图1)[2],反应的对映选择性高达99%。此反应一次性形成了两个C-C键、两个手性中心及一个季碳中心,可以用来合成一些手性的天然环丙烷类化合物。2008年,Cordova小组报道再次报道了α,β-不饱和醛与α-溴代丙二酸酯的不对称串联反应[3],取得了类似的结果。
图1 α,β-不饱和醛与α-溴代丙二酸酯的不对称串联反应
作者还研究了α-溴代乙酰乙酸乙酯与α,β-不饱和醛的不对称串联反应。在相同的反应条件下,得到了2-醛基环丙烷类化合物,该反应以非常高的非对映选择性(>25:1)和对映选择性(高达94%)得到了目标产物(图2)。
图2 α-溴代乙酰乙酸乙酯与α,β-不饱和醛的不对称串联反应
2007年,王伟小组也报道了α,β-不饱和醛与α-溴代丙二酸酯的不对称串联反应。该反应以3为催化剂,以2,6-lutidine为添加剂,顺利得到目标产物,并保持了反应优秀的对映选择性 (图3)[4]。

图3 α,β-不饱和醛与α-溴代丙二酸酯的不对称串联反应
作者经过进一步研究发现,使用乙酸钠为添加剂时得到了一种副产物(环丙烷的开环产物),作者认为底物8与9在催化剂作用下,首先发生Michael/α-烷基化反应,接着发生了脱质子与开环反应(retro-Michael)得到产物11 (图4)。

图4 脱质子与开环反应(retro-Michael)
与此同时,Ramon Rios小组报道了3催化的α,β-不饱和醛与不对称α-溴代丙二酸酯的不对称串联反应[5],得到了与Cordova小组相似的结果(图5)。该反应以非对称的α-溴代-β-酮酯为亲核试剂,得到的产物分子中含有一个季碳手性中心,反应的对映选择性高达99%。
图5 α,β-不饱和醛与不对称α-溴代丙二酸酯的不对称串联反应
2010年,Campagne小组报道了α-取代的α,β-丙烯醛与α-溴代丙二酸酯的不对称串联反应[6],成功地在目标产物分子中引入了一个手性季碳中心。作者对添加剂的影响进行了比较研究,发现以2,6-Lutidine 为添加剂比N-Methylimidazole得到的结果要好,其非对映选择性和对映选择性都有所提高。作者对得到的底物进行了衍生,获得了多手性中心的桥环内酰胺和桥环内酯(图6)。

图6 α-取代的α,β-丙烯醛与α-溴代丙二酸酯的不对称串联反应
2009年,鄢明小组报道了脱甲基奎宁20催化的α,β-不饱和硝基烯与α-溴代丙二酸酯的不对称串联反应[7]。通过添加DABCO,反应可以高选择性地得到反式结构的α-硝基环丙烷类化合物(图7)。
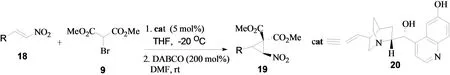
图7 α,β-不饱和硝基烯与α-溴代丙二酸酯的不对称串联反应
2010年,Lattanzi小组也报道了α,β-不饱和硝基烯与α-溴代丙二酸酯的不对称串联反应。该小组使用的催化剂为2,2’-(二萘基)脯氨醇21e,反应的对映选择性只有49% (图8)[8]。
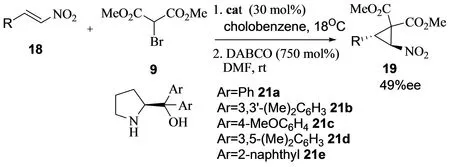
图8 α,β-不饱和硝基烯与α-溴代丙二酸酯的不对称串联反应
'2011年,Maruoka小组报道了在相转移催化剂作用下α-取代α,β-不饱和硝基烯烃与α-溴代丙二酸酯的不对称串联反应。该反应以23a为催化剂、碳酸铯为添加剂,以良好的对映选择性得到了S-构型的异噁唑啉类化合物(图9)[9]。该反应的一大优点是催化剂的用量降低到了1 mol %。
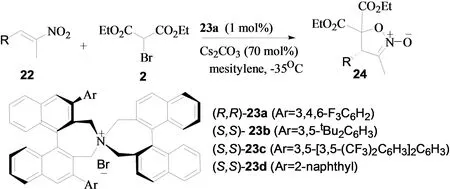
图9 α-取代α,β-不饱和硝基烯烃与α-溴代丙二酸酯的不对称串联反应
2011年,Lattanzi小组报道了α-芳基亚甲基-1,3-茚满二酮与α-溴代丙二酸酯的不对称串联反应[10]。在此反应中,作者发现2,2’-二(3,5-二甲基苯基)脯氨醇21d是最有效的催化剂(67% ee)。由于在反应过程中生成的溴化氢会引起催化剂的中毒失活,因此需要加入100 mol %的催化剂。此外,该反应还有一些不足,那就是反应的对映选择性不高,只获得中等到良好的对映选择性(图10)。
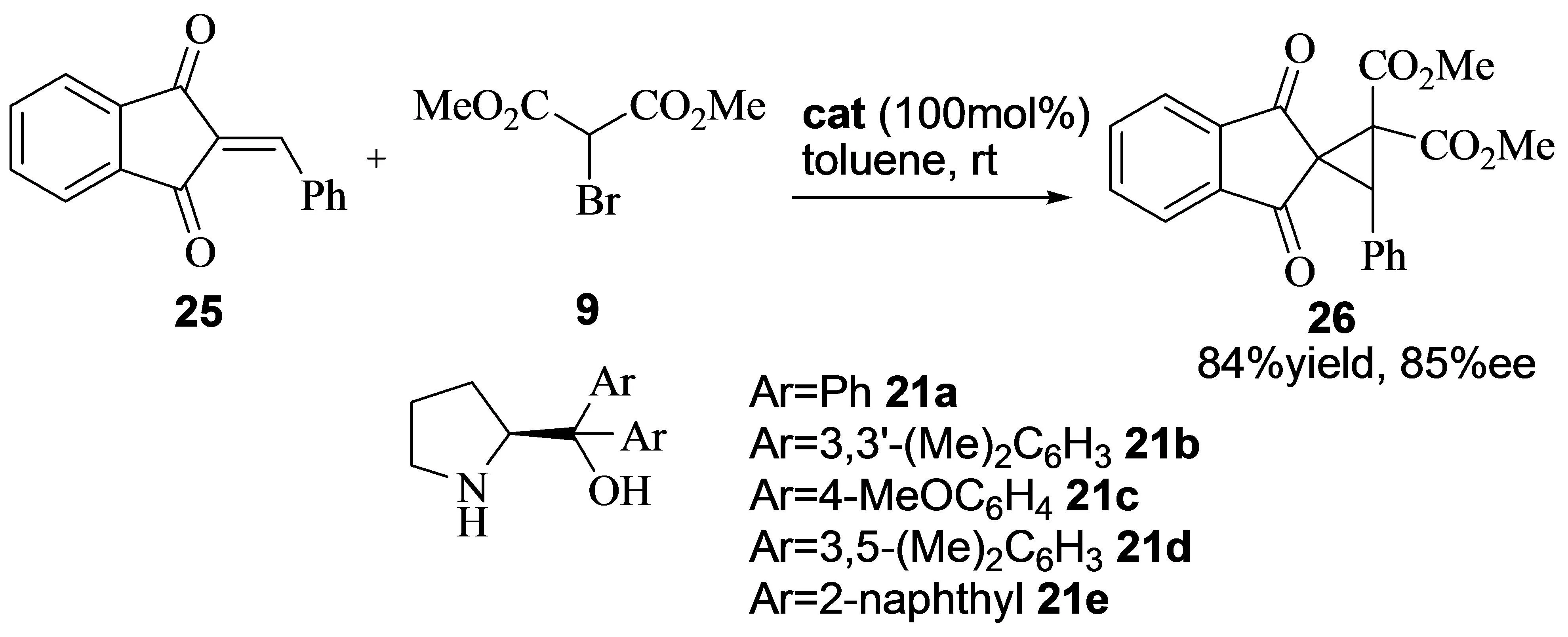
图10 α-芳基亚甲基-1,3-茚满二酮与α-溴代丙二酸酯的不对称串联反应
1.2 有机催化α-卤代硝基烷烃化合物的串联反应
α-卤代硝基甲烷是最具代表性的α-卤代硝基烷烃,不仅能与一些Michael受体发生串联反应得到硝基环丙烷类化合物[11],还能与亚胺发生串联反应得到具有生物活性的吖啶类化合物,这类反应具有广阔的应用前景。
2006年, Steven V. Ley小组报道了proline tetrazole 29催化的环己烯酮与溴代硝基甲烷的不对称串联反应[12],以中等到良好的对映选择性得到硝基环丙烷类化合物(图11)。作者考查了碱性添加剂对反应对映选择性的影响,发现以吗啡啉作为添加剂,反应的对映选择性最好。
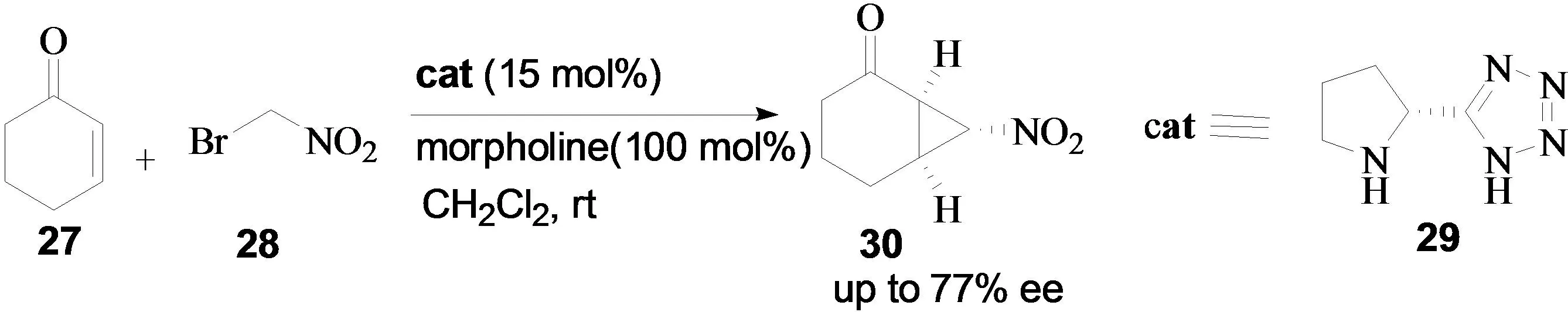
图11 环己烯酮与溴代硝基甲烷的不对称串联反应
2009年,鄢明小组也报道了环己烯酮与溴代硝基甲烷的不对称串联反应[13]。使用了另外一种催化剂(手性单磺酰二胺)31,取得了很好的实验结果。同时考查了酸/碱添加剂对反应的影响,发现酸添加剂和碱添加剂对反应的对映选择性都有着显著的影响:在没有加入酸添加剂的情况下,只能得到少量的产物;而在没有加入碱添加剂的情况下,却得不到目标产物。进一步研究发现,以苯甲酸和N-甲基吗啡啉(NMM)作为混合添加剂,反应得到了最好的实验结果,以90%的产率和95%的对映选择性得到目标产物(图12)。
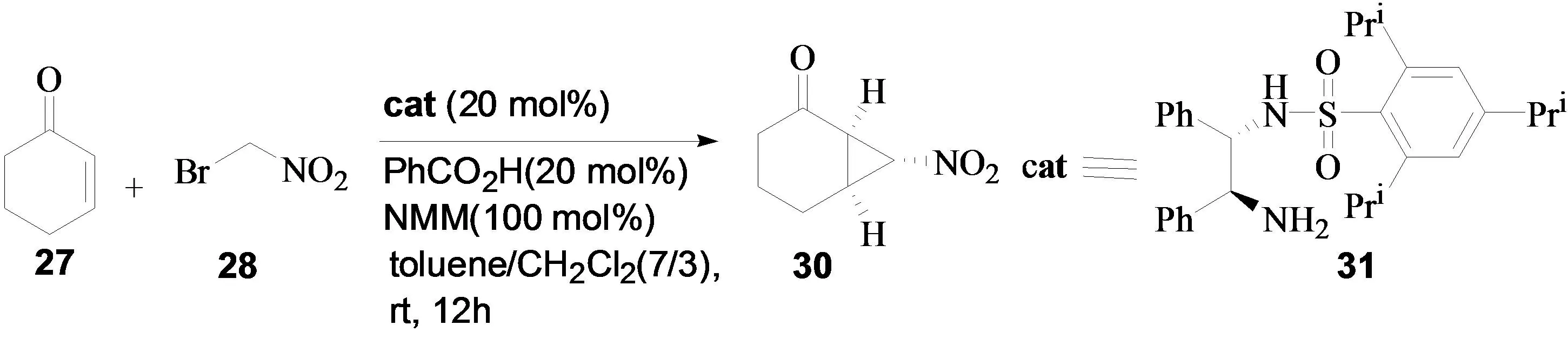
图12 环己烯酮与溴代硝基甲烷的不对称串联反应
与此同时, 王咏梅小组也报道了环己烯酮与溴代硝基甲烷的不对称串联反应[14]。在奎宁胺32的催化下,反应获得了非常优秀的产率和对映选择性。不但α-溴代硝基甲烷在该反应中取得了非常好的实验结果,其他的α-溴代硝基烷烃(如α-溴代硝基乙烷)在该类反应也能取得很好的实验结果(图13)。作者提出了反应的机理,在手性奎宁胺32的催化下,底物27和28首先反生不对称Michael反应形成过渡态,接着发生α-烷基化反应得到关环产物,比较合理的解释了反应现象及实验结果。
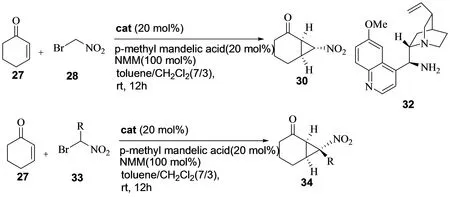
图13 环己烯酮与溴代硝基烷烃的不对称串联反应
2008年,Córdova小组报道了α,β-不饱和醛与溴代硝基甲烷的不对称串联反应[15]。作者对催化剂进行了筛选,发现3是最好的催化剂。作者进一步考查了碱添加剂对反应的影响,发现在以三乙胺作为碱添加剂时,反应得到了最好的实验结果(图14)。产物经过进一步的转化,得到了β-硝基羧酸酯,而β-硝基羧酸酯是合成药物Baclofen的前体,在其它的一些手性药物合成中也具有很高的应用价值。

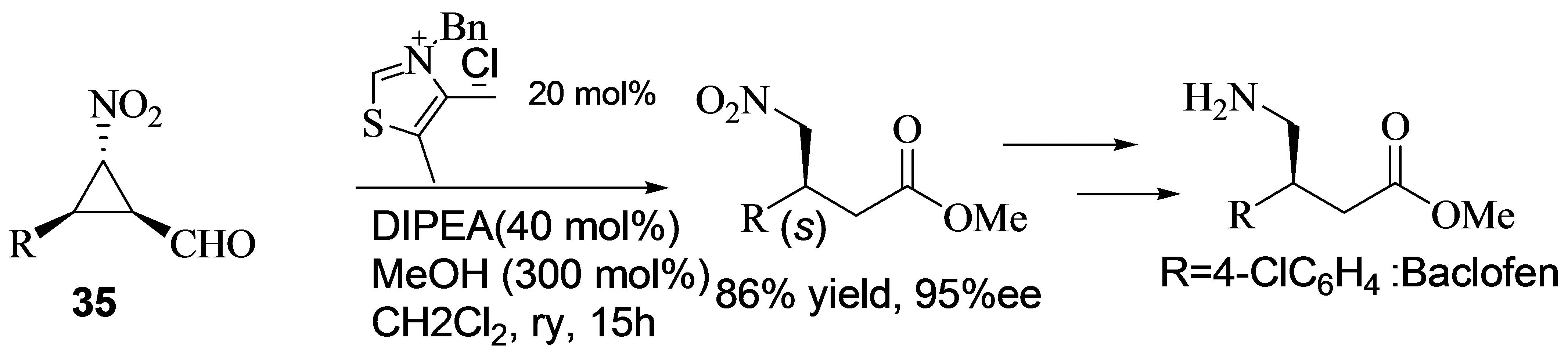
图14 α,β-不饱和醛与溴代硝基甲烷的不对称串联反应
与此同时, 鄢明小组也报道了α,β-不饱和醛与溴代硝基甲烷的不对称串联反应[16]。通过改变反应条件,在甲醇和乙酸钠的反应体系中,产率得到明显提高,而且产物的对映选择性也有所提高。此外,催化剂的用量降低到了10 mol % (图15)。
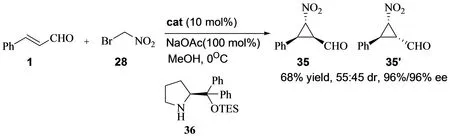
图15 α,β-不饱和醛与溴代硝基甲烷的不对称串联反应
2009年,Takemoto小组报道了α,β-不饱和-α-腈基酰亚胺与溴代硝基甲烷的不对称串联反应[17]。作者发现双功能硫脲是最有效的催化剂,通过对催化剂的筛选,确定环己二胺衍生的叔胺硫脲39给出最好的产率和对映选择性。同时考查了碱添加剂对反应的影响,发现三乙胺是最好的添加剂(图16)。

图16 α,β-不饱和-α-腈基酰亚胺与溴代硝基甲烷的不对称串联反应
2011年,Bencivenni小组报道了羟基吲哚类衍生物与溴代硝基甲烷的不对称串联反应[18]。通过对催化剂的筛选,作者发现奎宁胺衍生的硫脲42为最优的催化剂。以碳酸钠为碱添加剂和甲基叔丁基乙醚为溶剂,该反应以19:1的非对映选择性和98%的对映选择性得到螺环羟基吲哚类化合物。这类化合物可作为HIV-1的抑制剂,在药物研究中具有非常重要的应用。作者对反应的机理进行了讨论,认为硫脲上的氢和酰亚胺上的羰基之间形成的三个氢键对反应的立体选择性起到了非常关键的作用(图17)。
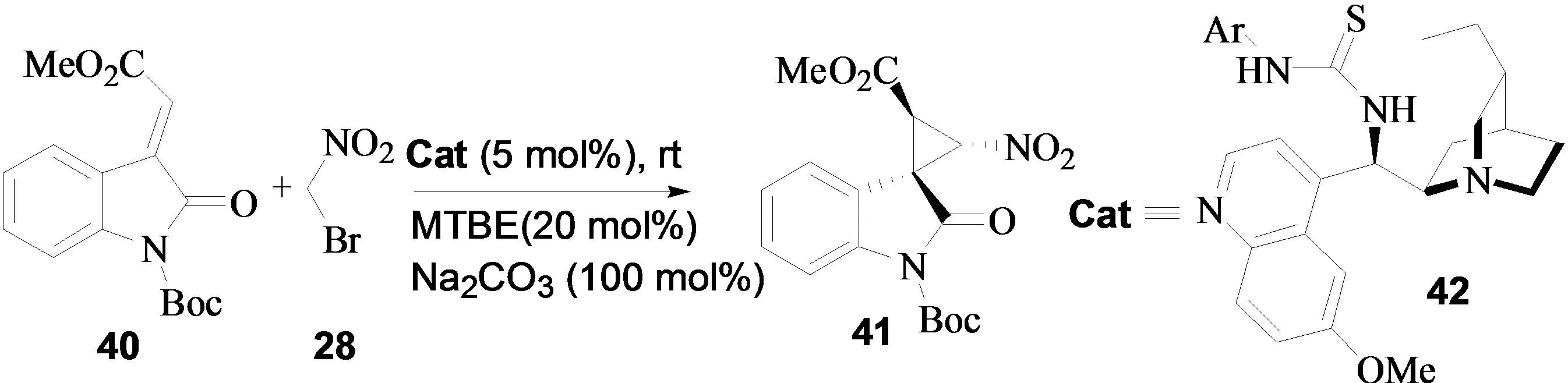
图17 羟基吲哚类衍生物与溴代硝基甲烷的不对称串联反应
采用亚胺类化合物作为亲电试剂与α-溴代硝基甲烷反应,可得到吖啶类化合物。吖啶广泛存在于多种天然产物及药物中[19],自然界中许多具有吖啶环的化合物表现出显著的生物活性,例如丝裂霉素A、B和C,还有泊飞霉素、丝紫霉素等,这些化合物都是从轮枝链霉菌的土壤浸出液中提取出来的重要天然抗肿瘤药物(图18)。吖啶环由于三元环的高度张力,可接受多种亲核试剂进攻,得到立体或区域选择性的开环产物,是合成含氮官能团化合物的有效中间体。

图18 具有吖啶环的几种抗肿瘤药物
2009年,Ritu Kapoor小组报道了磺酰亚胺与α-溴代硝基甲烷的串联反应,得到以顺式吖啶为主的产物。该类产物具有很高的合成利用价值,如果将来能发展为不对称催化反应,其利用价值会更高(图19)[20]。
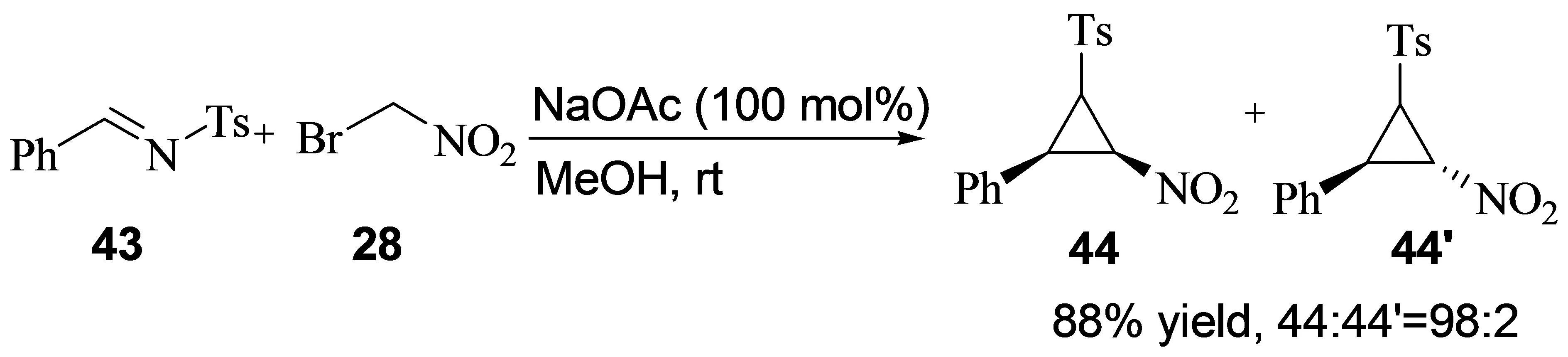
图19 磺酰亚胺与α-溴代硝基甲烷的串联反应
1.3 有机催化其它α-卤代亲核试剂的串联反应
α-卤代醛是典型的α-卤代亲核试剂,主要用于氮杂卡宾催化的反应中,如:Diels-Alder反应、[4+1]环加成反应。在2006年,Bode小组报道了手性氮杂卡宾催化的α-卤代醛与α,β-不饱和-γ-酮酯的Diels-Alder反应[21]。作者发现在手性催化剂(手性茚醇衍生的氮杂卡宾48)的催化下,该反应可以高对映选择性得到目标产物。同时,作者考查了碱添加剂对反应的影响,发现在三乙胺存在时,反应的结果最好(88% yield, >20:1 dr, 99% ee)。在此反应中添加剂的作用是中和反应过程中生产的HCl (图20)。
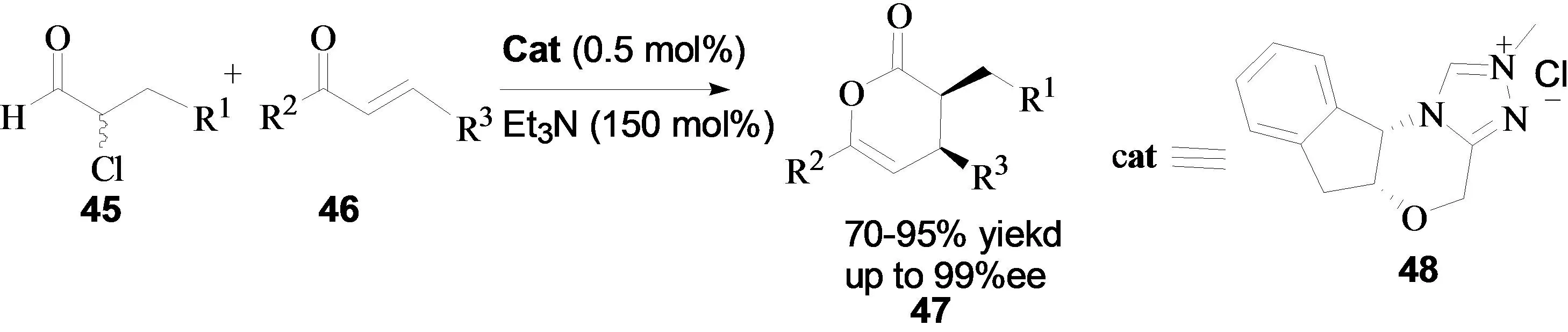
图20 α-卤代醛与α,β-不饱和-γ-酮酯的Diels-Alder反应
2011年,游书力小组报道了1,2-二苯基乙二胺衍生的手性卡宾52催化的α-卤代苯丙醛的不对称Diels-Alder反应[22]。作者也考查了碱添加剂对反应的影响,也发现三乙胺是最好的添加剂。该反应在催化剂52和三乙胺存在下,取得了最好的结果,以中等到良好的产率和对映选择性得到目标产物(图21)。
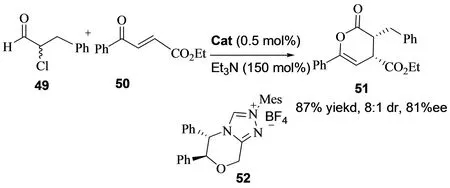
图21 α-卤代苯丙醛的不对称Diels-Alder反应
在2010年,钟国富小组报道了手性仲胺催化的α-卤代醛与硝基烯烃的[4+1]关环反应,得到了非常有用的顺式异唑啉类化合物[23]。作者考查了催化剂的影响,发现叠氮衍生的催化剂4e的催化效果最好。同时考察了α-卤代醛中卤素对反应的影响,发现底物为α-氯代醛时,得到的结果不理想。如果把底物改用α-溴代醛时,反应的产率和对映选择性有明显的提高。使用α-碘代醛能取得最好的实验结果(94% yield, 11:1 dr, >99% ee)。作者提出了反应的机理,认为在手性仲胺56的催化下,底物53和54首先反生不对称Michael反应,形成过渡态B。接着发生氧杂亲核取代反应得到关环产物(以顺式产物55为主),比较合理地解释了反应现象及实验结果(图22)。
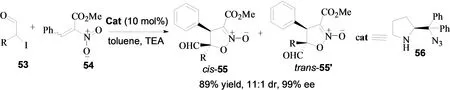
图22 α-卤代醛与硝基烯烃的[4+1]关环反应
2 有机催化的γ-卤代亲核试剂的串联反应
γ-卤代亲核试剂是一类很重要的卤代亲核试剂,广泛应用于串联反应中。但是,有机催化的γ-卤代亲核试剂参与的串联反应报道得很少。
2007年,Co′rdova报道了γ-溴代乙酰乙酸乙酯与α,β-不饱和醛的不对称串联反应[24]。在手性仲胺催化剂3的催化下,反应取得了最好的实验结果,其对映选择性高达98%(图23)。作者对反应机理进行了解释,认为手性仲胺与底物反生Michael加成反应得到中间体B,接着发生γ-烷基化得到环戊烷类化合物。这类化合物广泛存在于天然产物中,它们的环戊基结构是一些具有生理和药理活性的天然产物的关键骨架。作者进一步研究发现,如果把γ-溴代乙酰乙酸乙酯换成γ-氯代乙酰乙酸乙酯,则只能得到第一步Michael加成的产物,不能发生后续的关环反应。

图23 γ-溴代乙酰乙酸乙酯与α,β-不饱和醛的串联反应
2012年,卢一新小组报道了硝基烯烃与γ-溴代乙酰乙酸乙酯的不对称串联反应[25]。通过对催化剂的筛选,作者发现苏氨酸衍生的叔胺硫脲62是最好的催化剂。进一步的研究发现,碱添加剂对反应的产率和对映选择性有着非常显著的影响。加入碱添加剂后,反应的产率和对映选择性有了明显的提高。在苏氨酸衍生的叔胺硫脲62的催化作用下,该反应以90%的产率和90%的对映选择性得到特窗酸类化合物(图24)。
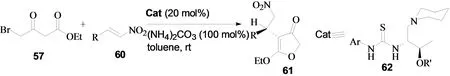
图24 硝基烯烃与γ-溴代乙酰乙酸乙酯的不对称串联反应
2011年,我们报道了4-氯代乙酰乙酸乙酯与亚胺的有机催化不对称Mannich加成反应[26]。通过对催化剂的筛选,发现基于吡咯烷的手性叔胺硫脲催化剂的催化性能最优。此外也对反应条件进行了优化,发现该催化体系对多种亚胺均能获得良好的催化活性和立体选择性。在此基础上,我们合成了一系列具有潜在生物活性的特窗酸类化合物(图25)。
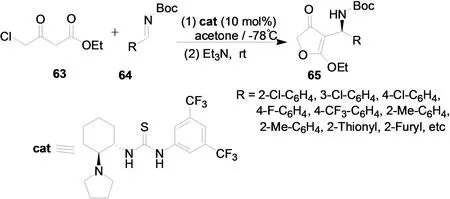
图25 亚胺与γ-氯代乙酰乙酸乙酯的不对称串联反应
3 总结与展望
有机催化的不对称串联反应已经成为当今有机合成中一类简便和高效的合成方法。众多的催化模式可以使科学家们充分地发挥创造力,设计出更有效的串联反应。虽然目前已经发展了很多种优秀的有机催化α-卤代亲核试剂的不对称串联反应,但有机催化的γ-卤代亲核试剂的串联反应报道很少。因此在以后的研究中,应该重视对有机催化γ-卤代亲核试剂的串联反应的研究,以合成更多有价值的手性分子。此外也需要发展催化效果更好的小分子催化剂,改进反应的收率和立体选择性。我们相信这个领域会在今后产生更多出色的结果,并且更广泛地应用于复杂天然产物和手性药物的合成。
[1] Grondal C, Jeanty M, Enders D. Organocatalytic cascade reactions as a new tool in total synthesis[J]. Nature Chemistry, 2010, 2 (3):167-178.
[2] Ramon R, Henrik S, Zhao G L, et al. A simple organocatalytic enantioselective cyclopropanation of α,β-unsaturated aldehydes [J]. Adv Synth Catal, 2007, 349: 1028 -1032.
[3]Ismail I, Zhao G L, Ramon R, et al. One-pot organocatalytic domino Michael/ α-alkylation reactions: direct catalytic enantioselective cyclopropanation and cyclopentanation reactions [J]. Chem Eur J, 2008, 14: 7867-7879.
[4] Xie H X, Zu L S, Li H, et al. Organocatalytic enantioselective cascade Michael-alkylation reactions: synthesis of chiral cyclopropanes and investigation of unexpected organocatalyzed stereoselective ring opening of cyclopropanes [J]. J Am Chem Soc, 2007, 129:10886-10894.
[5] Xavier C, Francisco C, Ramon R, et al. Asymmetric organocatalytic cyclopropanation-highly stereocontrolled synthesis of chiral cyclopropanes with quaternary stereocenters [J]. Eur J Org Chem, 2009, 77:3075-3080.
[6] Vincent T, Arievander L, Renata M, et al. Organocatalyzed cyclopropanation of a-substituted α,β-unsaturated aldehydes: enantioselective synthesis of cyclopropanes bearing a chiral quaternary center [J]. Chem Eur J, 2010, 16:7875-7880.
[7] Xuan Y N, Nie S Z, Yan M, et al. Highly enantioselective synthesis of nitrocyclopropanes via organocatalytic conjugate addition of bromomalonate to α, β-unsaturated nitroalkenes [J]. Org let, 2009, 11: 1583-1586.
[8]Alessio R, Alessandra L. Stereoselective synthesis of functionalised cyclopropanes from nitroalkenes via an organocatalysed Michael-initiated ring-closure approach [J]. Tetrahedron Asymmetry, 2010, 21: 1155-1157.
[9] Taichi K, Akihiro Y, Sunhwa S, et al. Catalytic asymmetric syntheses of isoxazoline-N-oxides under phase-transfer conditions [J]. Chem Commu, 2011, 47: 4358-4360
[10] Alessio R, Sara M, Consiglia T, et al. Synthesis of activated cyclopropanes by an MIRC strategy: anenantioselective organocatalytic approach to spirocyclopropanes [J]. Eur J Org Chem, 2011, 79:5096-5103.
[11] (a) Donaldson W A. Synthesis of clclopropane containing natural products. Tetrahedron, 2001, 57: 8589-8591; (b) Wessjohann L A, Brandt W, Thiemann T. Biosynthesis and metabolism of cyclopropane rings in natural compounds [J]. Chem Review, 2003, 103: 1625-1629..
[12] Henriette M, Hansen D A, Steven V L, et al. A new asymmetric organocatalytic nitrocyclopropanation reaction [J]. Chem Commun, 2006, 45:4838-4840.
[13] Du Q S, Dong L T, Yan M, et al. Asymmetric conjugate addition of bromonitromethane to cyclic enones catalyzed by chiral monosulfonated diamines [J]. Arkiivoc, 2009, (xiv) :191-199.
[14] Lv J, Lin Z, Wang Y M, et al. Enantioselective synthesis of functionalized nitrocyclopropanes by organocatalytic conjugate addition of bromonitroalkanes to α , β-unsaturated enones [J]. Chem Eur J, 2009, 15:972-979.
[15] Jan V, Zhao G L, Armando C. Organocatalytic asymmetric nitrocyclopropanation of α, β-unsaturated aldehydes [J]. Tetrahedron Letters, 2008, 49: 4209-4212.
[16] Zhang J M, Yan M, Hu Z P, et al. Organocatalytic conjugate addition of 1-bromonitroalkanes to α, β-unsaturated aldehydes: synthesis of nitrocyclopropanes [J]. Tetrahedron, 2009, 65: 802-806.
[17] Tsubasa I, Shota S, Yoshiji T. Enantioselective nitrocyclopropanation of α, β-unsaturated-α-cyanoimides catalyzed by bifunctional thiourea [J]. Syn lett, 2009, 2009:1627-1630
[18] Fabio P, Paolo R, Andrea M, Giorgio B. Organocatalytic Michael-alkylation cascade: the enantioselective nitrocyclopropanation of oxindoles [J]. Chem Eur J, 2011, 17:2842-2845.
[19] (a) Sweeney J B, Aziridines. epoxides' ugly cousins. Chem Soc Rev, 2002, 31 (5): 247-258; (b) Tanner D. Chiral aziridines-their synthesis and use in stereoselective transformations [J]. Angew Chem, 1994, 33(6): 599-619.
[20] Lal D S, Yadav G, Ritu K. The first diastereoselective nitroaziridination of N-tosylaldimines with 1-bromonitroalkanes [J]. Tetrahedron Letters, 2009, 50: 5420-5423.
[21] He M, Gerson J U, and Jeffrey W B. Chiral N-heterocyclic carbene catalyzed cnantioselective oxodiene Diels-Alder reactions with low catalyst loadings [J]. J Am Chem Soc, 2006, 128:15088-15089.
[22] Rong Z Q, Jia M Q, You S L. (1R,2R)-DPEN-derived triazolium salts for enantioselective oxodiene Dielse-Alder reactions [J]. Tetrahedron, 2011, 67: 9329-9333.
[23] Shi Z G, Tan B, Zhong G F, et al. Catalytic asymmetric formal [4+1] annulation leading to optically active cis-isoxazoline N-oxides [J]. Org let, 2010, 12:5402-5405.
[24] Ramon R, Jan V, Henrik S, et al.One-pot organocatalytic domino Michael/α-alkylation reactions: highly enantioselective synthesis of functionalized cyclopentanones and cyclopentanols [J]. Tetrahedron Letters, 2007, 48:5835-5839.
[25]Dou X W, Han X Y, Lu Y X. From the Feist-Benary reaction to organocatalytic domino Michael-alkylation reactions: asymmetric synthesis of 3-(2H)-furanones [J]. Chem Eur J, 2012, 18: 85-89.
[26] Luo N H, Sun X, Yan M, et al. Asymmetric synthesis of O-alkylated tetronic acid derivatives via an organocatalytic Mannich reaction and subsequent intramolecular cyclization [J]. Tetrahedron Asymmetry, 2011, 22:1536-1541.
Organocatalytic Cascade/domino Reactions of Halogented-nuclear Reagent
LUO Nian-hua1, ZHONG Yu-hong1, WANG Bo1, WANG Yong1, CAI Jiu-biao2
(1.School of Chemistry and Chemical Engineering, Shangrao Normal University, Shangrao Jiangxi, 334001; 2. The Third Middle School of Xin-jian, Nanchang Jiangxi, 330100)
Recently, great attention has been directed to the organocatalytic cascade reactions of nuclear reagent. Chemists have developed a large number of new value-cascade reactions and have prepared many useful chiral and drug molecules. The common nuclear reagents included α or γ- Halogenated nuclear reagents. Firstly, the nuclear reagents containing α or γ- Halogen reacted with the affinity reagent and then, reacted with the nuclear reagent to achieve the cascade reactions. This review summarizes recent advances of organocatalytic cascade reactions of α or γ- Halogenated nuclear reagents.
nuclear reagent; cascade/domino reactions; halogenated; organocatalytic
2015-06-23.
江西省教育厅科技计划项目( GJJ14717)
罗年华(1982-),男,江西吉安人,讲师,博士,主要研究方向为不对称合成和方法学研究。E-mail:luoxiaoge102@163.com
O626
A
1004-2237(2015)06-0063-10
10.3969/j.issn.1004-2237.2015.06.013
