茂金属-Ziegler-Natta催化剂协同催化丙烯聚合
2015-06-06许泽楠董金勇
牛 慧,许泽楠,董金勇
(1. 中国科学院 化学研究所 工程塑料重点实验室,北京 100190;2. 中国科学院大学,北京 100049;3. 大连理工大学 化工学院高分子材料系 精细化工国家重点实验室,辽宁 大连 116024)
茂金属-Ziegler-Natta催化剂协同催化丙烯聚合
牛 慧1,3,许泽楠1,2,董金勇1
(1. 中国科学院 化学研究所 工程塑料重点实验室,北京 100190;2. 中国科学院大学,北京 100049;3. 大连理工大学 化工学院高分子材料系 精细化工国家重点实验室,辽宁 大连 116024)
基于茂金属催化剂rac-CH2(3-tBu-1-Ind)2ZrCl2和以双醚化合物为内给电子体的MgCl2/TiCl4型Ziegler-Natta催化剂,以甲基铝氧烷(MAO)和三异丁基铝分别作为Zr和Ti两种催化剂组分的助催化剂,研究了Zr-Ti协同催化下的丙烯聚合,考察了丙烯压力、溶剂种类、两种催化剂比例等对聚合的影响;用1H NMR,GPC,SEM等手段分析聚合物的结构和组成。实验结果表明,将具有产生端基含乙烯基双键的聚丙烯大分子单体功能的茂金属催化剂和具有催化丙烯共聚功能的Ziegler-Natta催化剂组合后,在较低的丙烯聚合压力(0.2 MPa)下只得到大分子单体和线型聚丙烯的混合物,而在较高压力(0.4 MPa)下可获得支化聚丙烯,进一步增加Zr-MAO浓度可大幅提高大分子单体的转化率,聚合物的Mark-Houwink曲线呈长链支化特征,所得聚丙烯粒子具有良好的球形颗粒形态。
茂金属催化剂;Ziegler-Natta催化剂;协同催化;丙烯聚合;支化聚丙烯
高分子化合物特有的长链结构赋予了其不同于小分子有机物和无机物的诸多特性。与线型高分子相比,具有长链支化结构的聚合物可大幅增加分子链之间的缠结,对聚合物溶液、熔体以及凝固态的性能都有显著影响[1-2]。对于应用广泛的聚丙烯,引入长链支化结构可克服普通线型聚丙烯熔体强度低而导致的一些加工性能缺陷,是聚丙烯高性能化的重要手段之一[3]。
长链支化聚丙烯的合成方法主要有后改性法和催化聚合法两大类。以高温辐照和反应挤出等为代表的后改性方法通过射线辐照[1,4]、过氧化物引发-接枝[5-7]等方法在熔融的线型聚丙烯链上产生支化结构,具有操作简单、适于直接加工等优点,是目前工业上制备长链支化聚丙烯的主要技术。由于后改性法的自由基反应本质,反应过程中如何有效避免聚丙烯的降解和交联等副反应是该方法需要解决的难题[8]。催化聚合法是利用催化剂催化单体聚合来间接或直接地获得长链支化结构:可以先合成大分子单体,然后大分子单体与丙烯共聚得到长链支化结构[9],也可以在具有反应性侧基的聚丙烯上引发支链生长[10],或采用直接原位共聚的方法通过多种单体的组合[11-14]或多种催化剂的组合[15-17]来制备长链支化聚丙烯。Ye等[11-12]利用C2-对称结构的茂金属催化剂催化丙烯与α,ω-双烯烃(如1,7-辛二烯、1,9-癸二烯)共聚,Zhai等[13-14]利用C2-对称结构的茂金属催化剂催化丙烯与苯乙烯基-α-烯烃共聚,均获得了长链支化聚丙烯,但反应中需要控制交联反应的发生。Weng等[15-17]利用两类催化性能不同的茂金属催化剂(或非茂催化剂)结合组成协同催化体系,其中一个催化剂催化丙烯聚合生成末端含乙烯基的聚丙烯大分子单体,另一茂金属催化剂催化大分子单体与丙烯原位共聚,得到长链支化聚丙烯。协同催化方法仅使用丙烯一种单体,且可完全避免交联等副反应发生,无论从聚合物结构调控角度还是从工艺难易角度考虑,都是更理想的方法。
由于目前聚丙烯工业中广泛使用的催化剂仍是载体型高效Ziegler-Natta催化剂,协同催化方法的两种催化剂中若有一种为Ziegler-Natta催化剂,都将具有重要的现实意义。与此同时,载体型Ziegler-Natta催化剂对聚合物形态控制的优势也将使这一方法更加适应现代聚丙烯工业的需要。
本工作提出一种茂金属/Ziegler-Natta催化剂协同催化合成长链支化聚丙烯的方法。考察了丙烯压力、溶剂种类、两种催化剂比例等对聚合的影响,并利用1H NMR,GPC,SEM等手段分析了聚合物的结构和组成。
1 实验部分
1.1 主要试剂
丙烯:聚合级,中国石化北京燕山石油化工有限公司;三异丁基铝(TIBA):1.1 mol/L的甲苯溶液,百灵威公司;甲基铝氧烷(MAO):1.4 mol/ L的甲苯溶液,Albemarle公司,按文献[18]报道的方法除去三甲基铝(TMA),得到白色固体粉末,并重新配成1.4 mol/L的甲苯溶液,备用;TiCl4:化学纯,国药集团化学试剂有限公司;9,9-二(甲氧基甲基)芴(BMMF):化学纯,营口市向阳催化剂有限责任公司;正己烷、甲苯:分析纯,北京化学试剂公司,金属钠干燥回流后蒸出使用;茂金属催化剂rac-CH2(3-tBu-1-Ind)2ZrCl2(催化剂Ⅰ):参考文献[19]报道的方法合成。Ziegler-Natta催化剂MgCl2/TiCl4/BMMF(催化剂Ⅱ):参考文献[20]报道的方法合成。
1.2 聚合
丙烯聚合按以下步骤进行(以表2中聚合Run 7为例):常温下,在干燥的450 mL不锈钢反应釜中通入丙烯气体0.1 MPa,用注射器依次加入50 mL正己烷、1.1 mol/L TIBA甲苯溶液1.0 mL和1.4 mol/ L的MAO甲苯溶液0.7 mL,搅拌5 min后,加入1.0 mL催化剂Ⅰ(浓度为2×10-3mol/L)和18 mg催化剂Ⅱ,向反应釜中继续通入丙烯气体至0.2 MPa,并升温至60 ℃,开始聚合。反应30 min后,放空釜内残留丙烯气体,将聚合物倒入盐酸/乙醇溶液中终止反应,依次用乙醇、蒸馏水洗涤后,在60 ℃下真空干燥8 h,得到聚合物7.06 g。
1.3 聚合物的表征
1H NMR表征在Bruker公司DMX 300型核磁共振仪上进行,溶剂为氘代邻二氯苯,测试温度110 ℃。
聚合物熔融与结晶性能的测试在Perkin-Elmer公司DSC-7A型示差扫描量热仪上进行,被测试样在N2保护下升温至200 ℃,以去除热历史,再以10 ℃/min的速率降至50 ℃并恒定3 min后,以10 ℃/min的速率升温至200 ℃,考察聚合物的熔融过程。
聚合物相对分子质量及其分布的测定在Agilent公司PL-220型高温凝胶渗透色谱仪(GPC)上进行,采用3根PL gel Mixed-B(10 μm)型色谱柱串联,并配置示差、黏度和光散射检测器。溶剂为1,2,4-三氯苯,测试温度150 ℃,流量1.0 mL/min。
聚合物颗粒的形貌采用JEOL公司JSM-6700型扫描电子显微镜观察,加速电压5 kV,试样在测试前进行喷铂金处理。
Mark-Houwink(M-H)方程给出了聚合物溶液特性黏数[η]与聚合物相对分子质量(Mw)之间的关系为:[η]=KMwα。其中,K为常数。对于线型高分子,[η]-Mw的双对数坐标是一条直线,斜率为α;而具有相同Mw的支化高分子,其[η]比线型高分子小,因此与线型聚合物的M-H曲线相比,支化聚合物的M-H曲线向下偏离,这一特征常用来判定聚合物中是否存在长链支化结构[21-22,10,14]。
2 结果与讨论
将催化剂Ⅰ与以双醚化合物为内给电子体的催化剂Ⅱ组成“协同催化”体系,催化丙烯聚合:用催化剂Ⅰ生成端基含反应性双键的等规聚丙烯大分子单体,并由催化剂Ⅱ催化大分子单体与丙烯的原位共聚,获得含有长链支化结构的等规聚丙烯。上述聚合过程包含3个主要步骤(见图1):①催化剂Ⅰ/MAO催化丙烯聚合产生大分子单体;②大分子单体在催化剂Ⅱ/TIBA的活性中心配位;③大分子单体与丙烯共聚。
2.1 催化剂Ⅰ/MAO催化丙烯聚合
茂金属/MAO催化体系催化的丙烯聚合中,在不外加链转移剂(包括氢气和烷基铝)的条件下,主要存在活性中心向β-H的链转移(β-H消除)和向β-Me的链转移(β-Me消除)反应,得到亚乙烯基、乙烯基、异丁烯基等端基结构[23-25],通过改变催化剂结构和聚合条件,可控制上述链转移反应的相对强弱,从而达到调节聚合物端基结构的目的。在各种端基结构中,乙烯基由于聚合能力最强,最适合作为大分子单体。但大多数茂金属催化剂主要发生β-H消除而生成亚乙烯基末端,能高比例地生成乙烯基端基的催化剂相对较少[26]。本课题组在研究茂金属催化剂Ⅰ时发现,由于茂环3-位上的叔丁基位阻较大,使其具有高的β-Me消除的特点,且对丙烯聚合的催化活性高、立构规整控制力强。

图1 茂金属/Ziegler-Natta协同催化丙烯聚合制备长链支化聚丙烯的反应机理Fig.1 Mechanism for the preparation of long chain-branched polypropylene with the metallocene/Ziegler-Natta tandem catalysis.BMMF:9,9-bis(methoxymethyl)fl uorene.
本实验以除去TMA的MAO(下同)作为催化剂Ⅰ的助催化剂以消除丙烯聚合中向TMA的链转移,催化剂Ⅰ/MAO催化丙烯聚合所得聚合物的端基组成,反应条件和聚合结果见表1中的Run 1,聚合物的1H NMR谱图见图2a。分析聚合物的端基结构表明,催化剂Ⅰ/MAO生成的聚丙烯中含有乙烯基、亚乙烯基和异丁烯基3种不饱和端基结构。通过计算得到乙烯基端基占3种端基总和的77.5%(x),表明β-Me消除是主要的链转移方式。
在催化剂Ⅰ和催化剂Ⅱ组成的协同催化体系中,需要使用烷基铝(本实验使用的是TIBA)作为催化剂Ⅱ的助催化剂,因此考察TIBA对催化剂Ⅰ/MAO催化丙烯聚合的影响十分必要。催化剂Ⅰ/MAO催化丙烯聚合随TIBA含量变化的规律见表1中的Run 2~4。Kleinschmidt等[27]曾报道烷基铝(TMA或TEA)的加入会使活性中心失活,然而本实验使用的TIBA由于具有较大的空间位阻,从而可有效地避免与活性中心的络合而影响催化活性。1H NMR表征结果显示,在所得的聚合物中乙烯基仍为主要端基结构,由计算可知,乙烯基端基含量均在70%(x)以上。
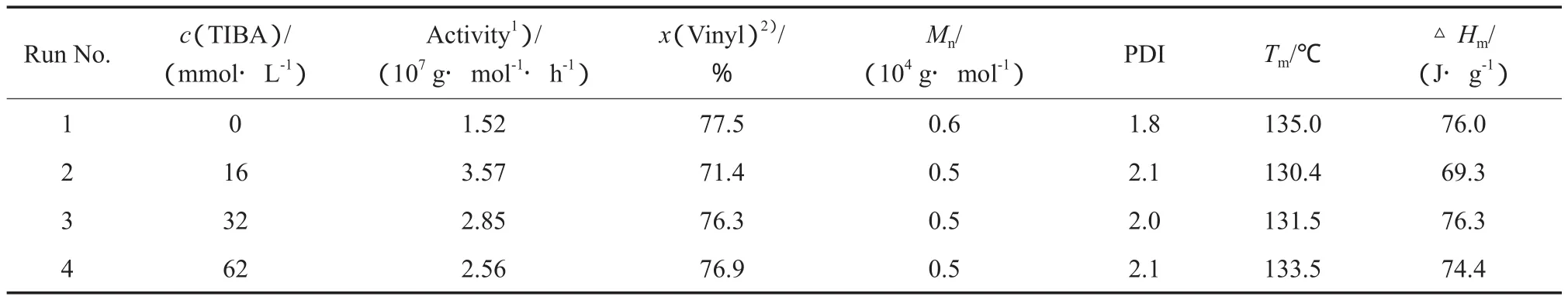
表1 TIBA对于催化剂Ⅰ/MAO催化丙烯聚合的影响Table 1 Effects of TIBA on the propylene polymerization catalyzed by catalystⅠ/MAO
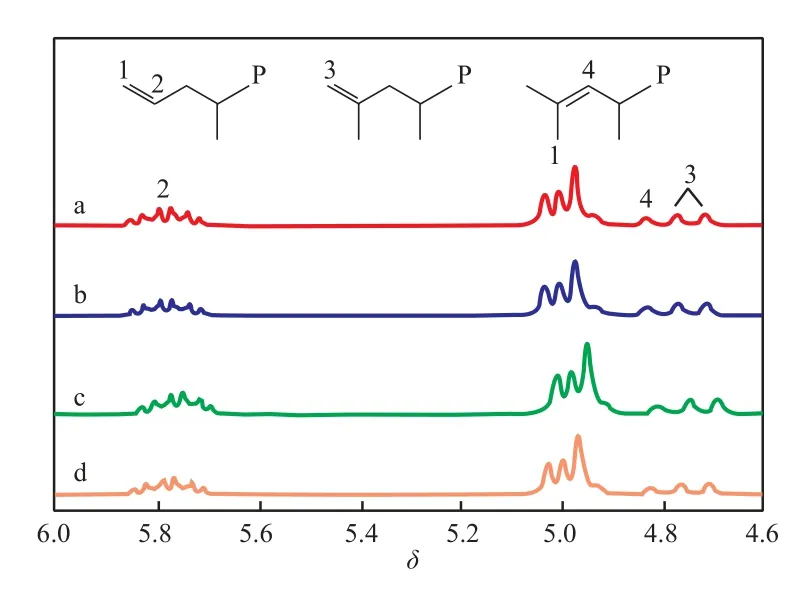
图2 催化剂Ⅰ/MAO和催化剂Ⅰ/MAO/TIBA催化得到的聚丙烯的1H NMR谱图Fig.21H NMR spectra of PP prepared with catalyst Ⅰ/MAO and catalyst Ⅰ/MAO/TIBA in Table 1.a Run 1;b Run 2;c Run 3;d Run 4
2.2 催化剂Ⅰ/MAO/催化剂Ⅱ/TIBA协同催化丙烯聚合
将催化剂Ⅰ/MAO和催化剂Ⅱ/TIBA组成具有协同催化的催化剂Ⅰ/MAO/催化剂Ⅱ/TIBA,用于丙烯聚合,使催化剂Ⅰ/MAO产生的聚丙烯大分子单体在催化剂Ⅱ/TIBA的催化下与丙烯单体进行共聚,通过改变催化剂Ⅰ/催化剂Ⅱ的比例、聚合压力、聚合温度及溶剂等反应条件,分别对大分子单体的浓度、相对分子质量以及溶解性等因素进行调节,来研究长链支化聚丙烯合成的影响因素。聚合条件和实验结果见表2。
聚合首先在60 ℃的正己烷溶剂中进行,丙烯气体压力设定为0.2 MPa,并与催化剂Ⅰ/MAO和催化剂Ⅱ/TIBA分别单独催化丙烯聚合的反应进行对比(Run 5和Run 6),协同催化方法得到的聚合物产量(Run 7)显著高于前两种催化剂单独催化得到的产量,表明催化剂Ⅰ和催化剂Ⅱ都有效地催化了丙烯聚合。所得聚合物的GPC曲线见图3a。由图3a可见,催化剂Ⅰ/MAO生成的大分子单体(Run 5)的相对分子质量较低,相对分子质量约为0.4×104g/mol,且相对分子质量分布较窄;催化剂Ⅱ/TIBA生成的聚丙烯(Run 6)相对分子质量较高,相对分子质量可达1.09×105g/ mol,相对分子质量分布宽;催化剂Ⅰ/MAO/催化剂Ⅱ/TIBA协同催化的聚合物7呈明显的双峰分布,其中,高相对分子质量部分为催化剂Ⅱ/ TIBA生成的聚丙烯,与催化剂Ⅱ/TIBA单独催化时相近,低相对分子质量部分为催化剂Ⅰ/MAO生成的聚丙烯,略高于催化剂Ⅰ/MAO单独催化的产物。聚合物的M-H曲线见图4a,催化剂Ⅱ/ TIBA催化丙烯聚合得到线型聚丙烯(聚合物6),其M-H曲线是一条直线。协同催化的聚合物7的M-H曲线在高相对分子质量末端出现非常轻微的向下偏离线性的趋势,表明在相同的相对分子质量下,聚合物7的[η]略小于聚合物6,这说明体系中生成了少量的支化结构。但聚合物7的GPC曲线中显著的双峰分布表明绝大多数大分子单体未参与共聚,支化聚合物的比例很低;且增加共聚催化剂的浓度(Run 8)亦不能明显促进大分子单体参与共聚。
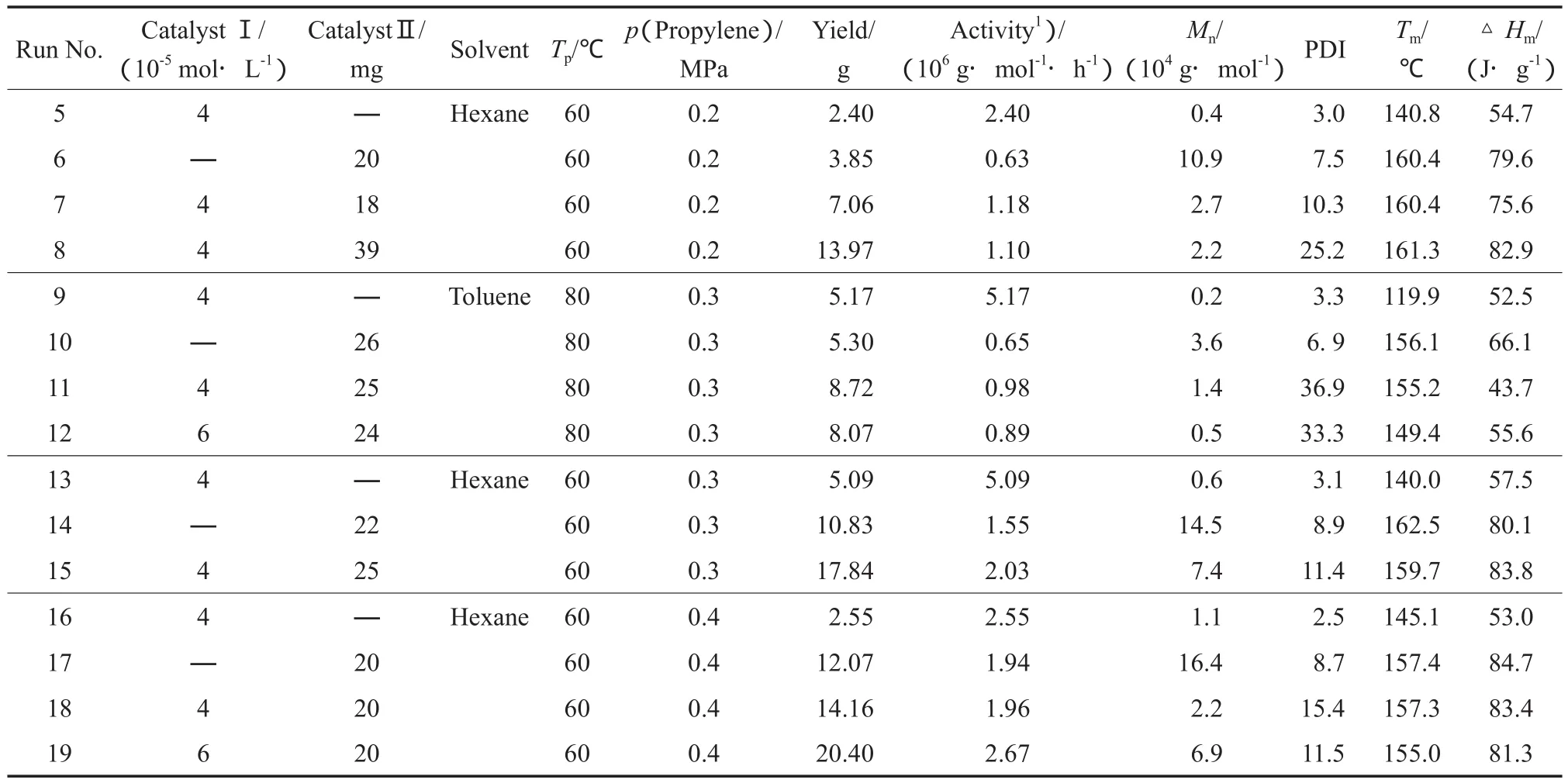
表2 催化剂Ⅰ/MAO/催化剂Ⅱ/TIBA协同催化下的丙烯聚合结果Table 2 Propylene polymerization catalyzed by catalystⅠ/MAO/catalystⅡ/TIBA tadem catalysis

图3 分别由催化剂Ⅰ/MAO、催化剂Ⅱ/TIBA和催化剂Ⅰ/MAO/催化剂Ⅱ/TIBA协同催化得到的聚丙烯的GPC曲线Fig.3 GPC curves of polymers prepared with catalystⅠ/MAO,catalystⅡ/TIBA and their tandem catalytic systems.a Run 5-8;b Run 9-12 and Run 15;c Run 16-19

图4 由催化剂Ⅱ/TIBA和催化剂Ⅰ/MAO/催化剂Ⅱ/TIBA协同催化得到的聚丙烯的M-H曲线Fig.4 Mark-Houwink curves of polymers prepared with catalystⅡ/TIBA and catalystⅠ/MAO/Ⅱ/TIBA tandem catalytic systems.a Run 6-8;b Run 10-12 and Run 14-15;c Run 17-19
如图1中的机理所示,大分子单体首先需要在非均相催化剂Ⅱ的活性中心上配位才能进行共聚,因此大分子单体靠近催化剂Ⅱ表面(尤其是内部表面)进而在Ti活性中心上配位是完成共聚的关键(见图1中的步骤②)。考虑到大分子单体在甲苯中具有更好的溶解性和运动能力,聚合Run 9~12在甲苯中进行,并通过升高温度(至80 ℃)来进一步促进大分子单体的溶解性和增加大分子单体的反应性。由于升温加速了链转移反应,催化剂Ⅰ/MAO得到的大分子单体(图3b中的Run 9)的相对分子质量降至0.2×104g/mol(为使大分子单体的相对分子质量不至于过低,将丙烯气体压力升至0.3 MPa)。协同催化方法得到的聚合物产量(Run 11)明显高于两种催化剂单独催化的产量(Run 9和Run 10),但低于两种催化剂的产量之和,表明两种催化剂均能催化丙烯聚合,但活性有所降低。由聚合物11的GPC曲线(见图3b)可见,虽然在曲线的低相对分子质量区域可见少量的大分子单体残留,但聚合物的主要组成仍是相对分子质量较高的聚丙烯;同时考虑到催化剂Ⅰ/MAO和催化剂Ⅱ/TIBA在该反应条件下催化丙烯聚合的产率相近(分别为5.17 g和5.30 g),因此可以判定大分子单体有很大部分参与了共聚而进入相对分子质量较高的聚丙烯部分(即由催化剂Ⅱ/TIBA催化了丙烯与大分子单体的共聚),生成了具有支化结构的聚丙烯。但聚合物11的M-H曲线(见图4b)仅在末端出现了轻微的偏离线性现象,整体部分与线型聚合物10基本重合,造成这一结果的原因可能是由于支链的相对分子质量太小(0.2×104g/mol)。增加大分子单体的含量(Run 12)并未提高聚合物的支化程度,反而由于聚合溶液黏度提高而使聚合物颗粒相互粘连,降低了催化剂Ⅱ的活性。由此可见,虽然使用甲苯作溶剂可提高大分子单体的溶解性和运动能力,但由于催化剂Ⅱ的非均相催化特质,聚合时还需考虑单体(包括大分子单体)在催化剂Ⅱ孔道内扩散的难易程度。对比Run 15可知,在溶解性适中的溶剂(正己烷)中,共聚活性显著增加,协同催化产率明显高于两种催化剂单独催化时的产量(Run 13和Run 14),且高于两者产量之和。通过聚合活性可粗略计算出催化剂Ⅰ产生的大分子单体约占总产物的1/3,而GPC曲线显示大分子单体残留量很低,说明大多数大分子单体参与了共聚。同时,聚合物的表观形态较甲苯中的产物有明显改善,聚合物的熔点也显著提高,这表明在正己烷溶剂中大分子单体向催化剂Ⅱ内部活性中心的扩散较其在甲苯中更顺畅。
对比Run 7和反应Run 15可发现,升高丙烯压力(丙烯单体浓度)对协同催化聚合物组成的影响显著。当丙烯压力从0.2 MPa增至0.3 MPa时,聚合物中残留大分子单体的比例可大幅减小,这一点通过对比聚合物7和聚合物15的GPC曲线可直观地看出。由于两个反应中的大分子单体相对分子质量均较低且差别不大(分别为0.4×104g/mol和0.6×104g/mol),本实验认为相对分子质量不应是造成大分子单体共聚能力差异的主要因素。由于MgCl2/TiCl4载体型Ziegler-Natta催化剂是具有多孔结构的非均相催化剂,在本研究中,催化剂Ⅱ生成的聚合物颗粒实际上成为大分子单体与丙烯单体共聚的“微反应器”[28],聚合压力的升高意味着每个“微反应器”颗粒可以长得更大且通过“形态复制”效应在颗粒内部产生更大和更多的孔隙,从而使大分子单体更容易运动至孔隙内的催化活性中心上,这对于图1中的步骤②是有利的。尽管聚合物15的M-H曲线并没有表现出明显的偏离线性行为,但根据上述对聚合活性和GPC曲线的分析可知,这并不是由于支化反应没有发生,而主要是由于支链的相对分子质量太低,对聚合物特性黏数的影响很小所致。
为进一步提高大分子单体的相对分子质量从而增加支链长度,Run 16将聚合压力提高至0.4 MPa,并将催化剂Ⅰ/MAO的[MAO]/[Zr]减至250,催化剂Ⅰ的聚合活性有所降低,但大分子单体的相对分子质量可增至1.1×104g/mol,熔点达到145 ℃。协同催化反应Run 18的聚合物产量明显高于两种催化剂单独催化时的产量。由聚合物18 的GPC曲线(见图3c)可见,有少量大分子单体残留,其M-H曲线可从相对分子质量1×105g/mol处分为A和B两段(见图4c),A段的特性黏数明显偏离线性,B段则与线型聚合物的M-H曲线重合。表明聚合物为两种结构的混合物:一种是相对分子质量较低而支化度较高的聚合物;一种是相对分子质量较高而支化度较低的聚合物。利用1H NMR对聚合物18进行表征,表征结果显示,聚合物中仍含有未反应的大分子单体,其不饱和端基结构清晰可见(见图5a)。Run 19将催化剂Ⅰ的浓度增至6×10-5mol/L,催化剂Ⅱ的加入量保持恒定,聚合活性和聚合物相对分子质量均明显增加。与Run 18相比,虽然增加了催化剂Ⅰ的浓度(即增加了大分子单体的产率),但聚合物的GPC曲线显示,在相对分子质量较低部分残存的大分子单体已非常少,且相对分子质量分布明显窄于其他的协同催化聚合物。聚合物19的M-H曲线在整个测试范围内均出现偏离线型聚合物的现象;随相对分子质量的增加,对线性曲线的偏离程度逐渐减弱,但在相对分子质量达1×107g/mol时,这种偏离仍清晰可辨。聚合物的1H NMR谱图中(见图5b)也已观察不到含有不饱和端基的大分子单体的存在。
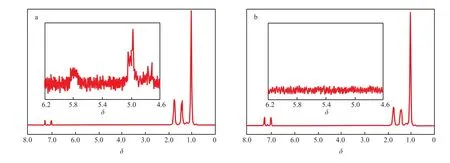
图5 催化剂Ⅰ/MAO/催化剂Ⅱ/TIBA协同催化得到的聚丙烯的1H NMR谱图Fig.51H NMR spectra of polymers prepared with the catalystⅠ/MAO/catalyst Ⅱ/TIBA tandem catalytic systems.a Run 18;b Run 19
由此可见,在此聚合条件(0.4 MPa)下,提高催化剂Ⅰ的浓度对于促进大分子单体参与共聚具有明显效果。这一现象可以用大分子单体在反应溶液中浓度分布的变化来解释。在本研究中,大分子单体由均相催化剂Ⅰ生成,其在聚合溶液中的溶解和分散是均一的,但只有催化剂Ⅱ附近的大分子单体容易在Ti活性中心上参与反应。提高催化剂Ⅰ的浓度(由于保持[MAO]/[Zr]恒定)相当于增加了MAO的浓度,在MAO与催化剂Ⅱ之间存在的Lewis酸-碱作用下,催化剂Ⅰ可在催化剂Ⅱ周围富集,进而使大分子单体在催化剂Ⅱ附近生成,甚至更多地在催化剂Ⅱ所产生的聚合物粒子的孔隙内部生成,这样大幅地提高了大分子单体共聚的转化率。利用SEM观察聚合物18和聚合物19的颗粒表观形态(见图6)可见,催化剂Ⅰ/MAO浓度较高时生成的聚合物颗粒具有更大的粒子直径、更完整的颗粒形态和更光滑的表面,表明共聚更多地在催化剂Ⅱ的颗粒内部进行。

图6 催化剂Ⅰ/MAO/催化剂Ⅱ/TIBA协同催化得到的聚丙烯颗粒的SEM照片Fig.6 SEM images of polymers prepared with the catalystⅠ/MAO/catalyst Ⅱ/TIBA tandem catalytic systems in Table 2.a Run 18;b Run 19
3 结论
通过研究催化剂Ⅰ和催化剂Ⅱ协同催化体系催化的丙烯聚合,探索了一种合成长链支化聚丙烯的方法。
1)催化剂Ⅰ/MAO可高效地产生含70%(x)以上乙烯基端基的聚丙烯大分子单体,催化剂Ⅱ/ TIBA则可催化丙烯与大分子单体原位共聚得到支化聚丙烯。
2)Ziegler-Natta催化剂生成的聚合物颗粒充当着共聚的“微反应器”,支化聚丙烯分子在“微反应器”内部的Ziegler-Natta催化剂活性中心上产生;通过升高聚合压力和增加茂金属-MAO络合物的浓度,可提高大分子单体的共聚转化率,增加支化高分子在聚合物中的比例。
3)通过简单的两种催化剂和助催化剂的一次性加入,得到了微观上具有长链支化结构、宏观上具有球形颗粒表观形态的支化聚丙烯。
[1] Auhl D,Stange J,Münstedt H,et al. Long-Chain Branched Polypropylenes by Electron Beam Irradiation and Their Rheological Properties[J]. Macromolecules,2004,37(25):9465 - 9472.
[2] Agarwal P K,Somani R H,Weng W,et al. Shear-Induced Crystallization in Novel Long Chain Branched Polypropylenes by in Situ Rheo-SAXS and WAXD[J]. Macromolecules,2003,36(14):5226 - 5235.
[3] 王红英,胡徐腾,李振宇,等. 高熔体强度聚丙烯的制备与表征[J]. 化学进展,2007,19(6):932 - 958.
[4] Valenza A,Piccarolo S,Spadaro G. Enhancement of Graft Yield and Control of Degradation During Polypropylene Maleation in the Presence of Polyfunctional Monomer[J]. Polymer,1999,40(4):835 - 841.
[5] Graebling D. Synthesis of Branched Polypropylene by a Reactive Extrusion Process[J]. Macromolecules,2002,35(12):4602 - 4610.
[6] Ni Qinglin,Fan Jiaqi,Niu Hui,et al. Enhancement of Graft Yield and Control of Degradation During Polypropylene Maleation in the Presence of Polyfunctional Monomer[J]. J Appl Polym Sci,2011,121(5):2512 - 2517.
[7] Wang Hongying,Guo Cunyue,Dong Jinyong. Catalytic Synthesis and Characterization of Well-Defi ned Polypropylene Graft Copolymers[J]. Catal Commun,2008,10(1):61 - 67.
[8] Chodák I,Zimányová E. The Effect of Temperature on Peroxide Initiated Crosslinking of Polypropylene[J]. Eur Polym J,1984,20(1):81 - 84.
[9] Weng Weiqing,Markel E J,Dekmezian A H. Synthesis of Long-Chain Branched Propylene Polymers via Macromonomer Incorporation[J]. Macromol Rapid Commun,2001,22(18):1488 - 1492.
[10] Wang Lu,Wan Dong,Zhang Zhenjiang,et al. Synthesis and Structure_Property Relationships of Polypropylene-g-Poly (Ethylene-co-1-Butene) Graft Copolymers with Well-Defi ned Long Chain Branched Molecular Structures[J]. Macromolecules,2011,44(11):4167 - 4179.
[11] Ye Zhibin,AlObaidi F,Zhu Shiping. Synthesis and Rheological Properties of Long-Chain-Branched Isotactic Polypropylenes Prepared by Copolymerization of Propylene and Nonconjugated Dienes[J]. Ind Eng Chem Res,2004,43(11):2860 -2870.
[12] Paavola S,Saarinen T,Löfgren B,et al. Propylene Copolymerization with Non-Conjugated Dienes and α-Olefi ns Using Supported Metallocene Catalyst[J]. Polymer,2004,45(7):2099 - 2110.
[13] Zhai Wentao,Wang Hongying,Yu Jian,et al. Foaming Behavior of Isotactic Polypropylene in Supercritical CO2Influenced by Phase Morphology via Chain Grafting[J]. Polymer,2008,49(13/14):3146 - 3156.
[14] Langston J A,Colby R H,Chung T C M,et al. Synthesis and Characterization of Long Chain Branched Isotactic Polypropylene via Metallocene Catalyst and T-Reagent[J]. Macromolecules,2007,40(8):2712 - 2720.
[15] Weng W,Hu W,Dekmezian A H,et al. Long Chain Branched Isotactic Polypropylene[J]. Macromolecules,2002,35(10):3838 - 3843.
[16] Cherian A E,Lobkovsky E B,Coates G W. Synthesis of Allyl-Terminated Syndiotactic Polypropylene:Macromonomers for the Synthesis of Branched Polyolefins[J]. Macromolecules,2005,38(15):6259 - 6268.
[17] Shiono T,Azad S M,Ikeda T. Copolymerization of Atactic Polypropene Macromonomer with Propene by an Isospecific Metallocene Catalyst[J]. Macromolecules,1999,32(18):5723 - 5727.
[18] Pédeutour J N,Radhakrishnan K,Cramail H,et al. Use of “TMA-Depleted” MAO for the Activation of Zirconocenes in Olefi n Polymerization[J]. J Mol Catal A:Chem,2002,185(1/2):119 - 125.
[19] Resconi L,Balboni D,Baruzzi G,et al. rac-[Methylene (3-tert-Butyl-1-Indenyl)2]ZrCl2:A Simple,High-Performance Zirconocene Catalyst for Isotactic Polypropene[J]. Organometallics,2000,19(4):420 - 429.
[20] 中国科学院化学研究所. 烯烃聚合载体催化剂体系及制备法:中国,1110281 A[P]. 1995 - 10 - 18.
[21] Zimm B H,Stockmayer W H. The Dimensions of Chain Molecules Containing Branches and Rings[J]. J Chem Phys,1949,17(12):1301 - 1314.
[22] Zimm B H,Kilb R W. Dynamics of Branched Polymer Molecules in Dilute Solution[J]. J Polym Sci,1959,37(131):19 -42.
[23] Resconi L. On the Mechanisms of Growing-Chain-End Isomerization and Transfer Reactions in Propylene Polymerization with Isospecifi c,C2-Symmetric Zirconocene Catalysts [J]. J Mol Catal A:Chem,1999,146(1/2):167 - 178.
[24] Kawahara N,Kojoh S,Matsuo S,et al. Study on Chain End Structures of Polypropylenes Prepared with Different Symmetrical Metallocene Catalysts[J]. Polymer,2004,45(9):2883 - 2888.
[25] Moscardi G,Resconi L,Cavallo L. Propene Polymerization with the Isospecific,Highly Regioselective rac-Me2C(3-t-Bu-1-Ind)2ZrCl2/MAO Catalyst:Ⅱ. Combined DFT/MM Analysis of Chain Propagation and Chain Release Reactions [J]. Organometallics,2001,20(10):1918 - 1931.
[26] Brintzinger H H,Fischer D,Mülhaupt R,et al. Stereospecifi c Olefi n Polymerization with Chiral Metallocene Catalysts[J]. Angew Chem,Int Ed Eng,1995,34(11):1143 - 1170.
[27] Kleinschmidt R,Leek Y,Reffke M,et al. Kinetics and Mechanistic Insight into Propylene Polymerization with Different Metallocenes and Various Aluminium Alkyls as Cocatalysts [J]. J Mol Catal A:Chem,1999,148(1/2):29 - 41.
[28] Cecchin G,Morini G,Pelliconi A. Polypropene Product Innovation by Reactor Granule Technology[J]. Macromol Symp,2001,173(1):195 - 210.
(编辑 李明辉)
敬告读者:《石油化工》自2015年第5期开始在“专题报道”栏目连续刊登中国石化北京化工研究院乙烯研究室的系列报道。主要针对乙烯研究室在裂解技术、数值模拟技术、抑制结焦技术、选择加氢技术、甲烷化技术、烯烃产品净化技术以及新型催化工艺开发与应用等方面的领先技术成果进行报道。敬请广大读者给予关注。
专题报道:中国石化北京化工研究院乙烯研究室基于图论算法实现了以正戊烷为原料,自动构建热裂解反应网络的模型程序。通过阐述反应网络数据结构的设计及反应网络自生成的实现,利用此模型构建的反应网络对实验工况进行模拟,在氢气、甲烷、乙烯和丙烯等主要产物的收率方面,相比手工构建的正戊烷热裂解自由基反应网络,自生成的正戊烷热裂解反应网络的模拟结果更为准确,具有明显优势。见本期669-673页。
中国石化北京化工研究院乙烯研究室简介:中国石化北京化工研究院乙烯研究室自20世纪60年代开始,长期致力于乙烯技术的研究和开发,围绕石油化工的“龙头”——低碳烯烃的生产和分离过程,先后完成了裂解炉辐射段工艺技术、裂解炉强化传热技术、裂解炉抗结焦涂层技术、裂解炉快速烧焦技术、选择加氢催化剂及技术、低温甲烷化催化剂及技术、超重机脱硫技术等核心技术的研发和工业应用。乙烯研究室裂解技术团队在对国外先进技术深入研究和消化吸收的基础上坚持创新发展,作为CBL裂解炉开发组的核心成员成功开发了我国首台20 kt/a裂解炉,随后裂解炉的产能实现了从60 kt/a、100 kt/a到150 kt/a的跨越式发展,目前采用CBL技术设计和改造裂解炉125台,总产能约为7 000 kt/a;与此同时,自主开发的强化传热技术、炉管抗结焦涂层的成功应用,使得国产化的裂解炉运行周期从50 d左右延长至200 d以上;乙烯研究室加氢催化剂技术团队通过不断创新,采用多种国际首创技术,开发了国内乙烯装置各种不同工艺技术所需的全部催化剂(应用于7种不同工艺与物料,共计十余个牌号),在催化剂性能等许多方面超越了国外同类催化剂,突破了国外大公司的垄断并迅速占领了国内80%以上的市场,表现出优异的增产节能、增收节支能力,取得了显著的经济效益和社会效益。技术上的领先,让我国自主研发的裂解炉和选择加氢催化剂成功走出国门。CBL裂解炉在马来西亚Titan公司成功开车,碳二、碳三选择加氢催化剂先后在英国、韩国、日本、伊朗、印度尼西亚、菲律宾、马来西亚、泰国、印度、沙特阿拉伯等国的石化企业成功应用。经过多年的努力,乙烯研究室在乙烯技术领域获得国家奖励5项。这些成果标志着中国石化的乙烯技术已达到国际先进水平,获得国际公司的认可。
Propylene Polymerization Catalyzed by Metallocene/Ziegler-Natta Tandem Catalysis
Niu Hui1,3,Xu Zenan1,2,Dong Jinyong1
(1. CAS Key Laboratory of Engineering Plastics,Institute of Chemistry,Chinese Academy of Sciences,Beijing 100190,China;2. University of Chinese Academy of Sciences,Beijing 100049,China;3. State Key Laboratory of Fine Chemicals,Department of Polymer Science and Engineering,School of Chemical Engineering,Dalian University of Technology,Dalian Liaoning 116024,China)
A new method for the synthesis of branched polypropylene was proposed using the tandem systems of metallocene(rac-CH2(3-tBu-1-Ind)2ZrCl2) and Ziegler-Natta(MgCl2/TiCl4/diether),in which methylaluminoxane(MAO) and triisobutylaluminium were used as cocatalysts for the Zr and Ti species respectively. The infl uences of propylene pressure,solvent types and Zr/Ti ratio on the polymerization were investigated and the polymers were characterized by means of1H NMR,GPC and SEM. The results revealed that after the metallocene catalyst,which was capable of promoting the function of macromonomer containing vinyl end-groups,was combined with the Ziegler-Natta catalyst which could catalyze the copolymerization of propylene,branched polypropylene was prepared under relatively high pressure (0.4 MPa of propylene),while a mixture of macromonomer and linear polypropylene were obtained under relatively low pressure(0.2 MPa of propylene). By increasing the Zr-MAO concentration the conversion of the macromonomer was remarkably improved in the copolymerization and a long chain-branching character was observed in the Mark-Houwink curves of the polymers. Moreover,the prepared polymers had good spherical granular morphology.
metallocene catalyst;Ziegler-Natta catalyst;tandem catalysis;propylene polymerization;branched polypropylene
1000 - 8144(2015)06 - 0674 - 09
TQ 325.1
A
2015 - 04 - 09;[修改稿日期] 2015 - 04 - 17。
牛慧(1977—),女,山西省太原市人,博士,副教授,电话 0411 - 84986482,电邮 hniu@dlut.edu.cn。联系人:董金勇,电话 010 - 82611905,电邮 jydong@iccas.ac.cn。
国家自然科学基金项目(51003105)。
