梗阻性黄疸伴肾功能不全
2015-06-01严雪敏敦志娜李景南陆星华
严雪敏,敦志娜,李景南,陆星华
1中国医学科学院北京协和医学院北京协和医院消化内科,北京1007302北京市怀柔医院消化内科,北京101400
梗阻性黄疸伴肾功能不全
严雪敏1,敦志娜2,李景南1,陆星华1
1中国医学科学院北京协和医学院北京协和医院消化内科,北京1007302北京市怀柔医院消化内科,北京101400
梗阻性黄疸;肾功能不全;IgG4相关性疾病
患者,男,74岁,因“上腹不适1月余,皮肤黄染22 d”入院。入院前1月余,患者无明显诱因出现进食后上腹不适伴纳差、厌油、恶心及乏力,否认明显发热、反酸、呕吐、腹痛、腹泻及尿量减少,未诊治。10 d后服用小诊所配制的含大黄成分的中药制剂后出现水样便1次,约500 ml,之后突发全身皮肤黄染伴瘙痒,可见陶土便及深色尿,否认发热、腰痛、腹痛、尿量减少、心悸等不适。本院门诊查肝肾功能示血清白蛋白 (albumin,Alb)35 g/L,谷丙转氨酶(alanine transaminase,ALT)99 U/L,谷草转氨酶(aspartate aminotransferase,AST)78 U/L,碱性磷酸酶 (alkaline phosphatase,ALP)303 U/L,谷氨酰转肽酶 (gamma-glutamyl transpeptidase,GGT)316 U/L,总胆红素 (total bilirubin,TBil)469.4 μmol/L,直接胆红素 (direct bilirubin,DBil)352.7 μmol/L,总胆汁酸 (total bile acid,TBA)118.0 μmol/L;肌酐(creatinine,Cr)355 μmol/L,尿素氮 (blood urea nitrogen,BUN)16.55 mmol/L;超声提示肝内外胆管扩张,胰腺因肠气干扰显示不清。考虑“梗阻性黄疸,肾功能不全”收入院。自发病以来,患者精神、食欲差,每2~3天排1次成形便,色偏浅;排尿1200~2000 ml/d,色深。否认尿频、尿急、尿痛。体重下降16.5 kg。高血压病史5年,血压最高150/110 mm Hg (1 mm Hg=0.133 kPa),未进行药物治疗及血压监测。多年不查体,自述平素健康无不适。妻患肺结核,5年前亡于肺癌。个人史、家族史无特殊。查体:血压150/90 mm Hg,皮肤、巩膜明显黄染;心肺腹无明显阳性体征。
上腹不适伴黄疸的主诉提示,患者除部分全身性疾病外,病变可能存在于肝胆胰等重要脏器。老年男性,典型的无痛性梗阻性黄疸,超声提示肝内外胆管扩张,应考虑壶腹周围癌。但因肠气影响超声未见明确胰头、胆管下段占位,应进一步完善血胰功、胰胆肿瘤标志物及壶腹周围脏器影像学检查。患者病程中明确提出服用小诊所中药后出现黄疸,并进行性加重,需考虑药物性肝损伤可能,但患者不伴发热及腹痛,为不支持点;患者是否存在其他基础肝病,并不清楚,需进一步完善肝功能评估。肾功能不全无法用高龄解释,病程中仅有1次大量腹泻,且总量仅500 ml,无法用肾前性因素解释。患者血压高,未正规治疗,是否存在长期未控制高血压而导致肾功能不全因素?但超声未提示肾脏缩小或皮髓质异常,为不支持点。
因肾功能不全,检查时应尽量避免使用含碘静脉造影剂。
进一步检查便常规+潜血 (-);肝功能示TBil 232.2 μmol/L↑,DBil 172.3 μmol/L↑,总白蛋白 (total albumin,TP)87 g/L,Alb 30 g/L↓,ALT 54 U/L (最高218 U/L),AST 49 U/L↑ ,ALP 262 U/L↑,GGT 162 U/L↑,TBA 58.9 μmol/L↑,乳酸脱氢酶(lactate dehydrogenase,LDH)216 U/L;凝血时间未见异常;血清蛋白电泳示Alb%40.8%↓,α1 2.8%,α2 7.1%,β1 3.0%↓,β2 7.8%↑,γ 38.5%↑;甲肝、乙肝、丙肝、戊肝病毒检测均阴性,巨细胞病毒、EB病毒-IgM均阴性、降钙素原 (procalcitonin,PCT)正常;胰功未见异常;红细胞沉降率 (erythrocyte sedimentation rate,ESR)105 mm/h↑;超敏C反应蛋白(high sensitivity C reactive protein,hsCRP)5.57 mg/L↑。血清肿瘤标志物CA19-9 87.7 U/ml,癌胚抗原 (carcinoembryonic antigen,CEA)、CA242、甲胎 蛋白(α-fetoprotein,AFP)等均正常。腹部 CT平扫及磁共振胰胆管成像 (magnetic resonance cholangiopancreatography,MRCP)提示胰头部胆总管狭窄,肝内胆管及胆总管上段扩张;胰腺形态饱满 (图1,2)。
患者辅助检查除外常见病毒性肝炎可能,无明显肝硬化证据,但血清蛋白电泳γ异常增高。高 γ球蛋白血症分为单克隆型和多克隆型,前者常见于骨髓瘤和淋巴瘤,后者多见于肝病、感染性疾病及免疫系统疾病。进一步行免疫球蛋白及免疫系统相关抗体检查。
患者胰功正常,仅CA19-9轻度升高,余肿瘤标志物均正常,CT平扫及MRCP均未无肝脏及胰腺存在占位证据,胰腺形态虽饱满如腊肠样,不除外胰腺炎,但胰周却无明显渗出,不似常见胰腺炎征象。胰头部胆总管狭窄可能为梗阻性黄疸的原因。

图1腹部CT平扫示胰腺肿大 (箭头)

图2磁共振胰胆管成像示胰头部胆总管狭窄 (箭头)
进一步完善各项实验室检查,包括:(1)免疫球蛋白检查:IgG 45.60 g/L↑,IgA、IgM(-);(2)补体及自身抗体等检查:C4 0.067 g/L,CH50、C3、抗链球菌溶血素O(anti-streptolysin O,ASO)、类风湿因子 (rheumatoid factor,RF)(-);抗核抗体 (antinuclear antibody,ANA)-IF(+)H 1∶320;抗双链DNA抗体 (抗ds-DNA抗体)、抗Ro 52抗体、抗多发性肌炎-硬皮病 (polymyositis scleroderma,PM-Scl)抗体、抗线粒体抗体 (anti-mitochondrial antibody,AMA)-M2、抗Sm(WB)、抗干燥综合征抗原A抗体(Sjgren's syndrome A antigen antibody,SSA)、抗SSB (WB)、Scl-70、Jo-1(WB)(-)。
ANA-IF(+)H 1∶320,提示患者有免疫性疾病倾向。IgG明显升高,应进一步完善IgG亚类分型。
IgG亚类:IgG1 15 100 mg/L↑,IgG2 6790 mg/L↑,IgG3 563 mg/L,IgG4 35 600 mg/L↑。
结合老年患者,ANA-IF(+)H 1∶320,IgG明显升高,影像学无恶性肿瘤提示,胰腺形态饱满无占位,需考虑自身免疫性胰腺炎 (autoimmune pancreatitis,AIP)可能;患者IgG4明显增高,考虑AIP 1型。
AIP为少见的胰腺良性疾病,1995年由Yoshida等[1]提出。本病特征为血清IgG4水平升高,是以胰腺实质内大量IgG4阳性淋巴-浆细胞组织浸润为特征的胰腺纤维炎症性疾病,伴有胰腺内外分泌功能障碍。AIP的临床表现为体重下降、黄疸及慢性非特异性腹痛,腹痛程度较轻。迄今尚无统一的诊断标准 (表1,2),随着研究深入,根据病理区分为1型和2型 (表3,4)。
可以看出,血清IgG4水平和组织学检查对诊断十分重要。
此外,患者胰头部胆总管狭窄为此次黄疸的原因,可能为肿大胰腺压迫,也可能为胆管自身病变(炎症或肿瘤)。首先应完善无创内镜下超声检查(endoscopic ultrasonography,EUS),必要时应行 EUS下病理活检或内镜下逆行胰胆管造影 (endoscopic retrograde cholangiopancreatography,ERCP)下活检术。
内镜下超声检查:胰腺增大,长径约3.3 cm,内部回声不均,未见明确占位病变;胆总管明显扩张,下段管壁明显均匀增厚;胆总管增宽 (1.1 cm),胰腺周围可见多发曲张静脉 (图3)。

表1自身免疫性胰腺炎亚洲诊断标准 (日韩标准)[2]

表2自身免疫性胰腺炎Mayo临床中心临床诊断标准:HISOR标准[2]

表3自身免疫性胰腺炎1型 (IgG4相关硬化性胰腺炎)PDSHOR分型[3]

表4自身免疫性胰腺炎2型 (IgG4相关硬化性胰腺炎)PDHOR分型[3]
EUS未发现明确壶腹、胰腺、胆管占位,基本除外恶性疾病。但胆总管下段明显增厚,结合患者血清IgG4明显升高,考虑存在 IgG4相关性疾病 (IgG4-related disease,IgG4-RD),AIP 1型,IgG4相关性胆管炎 (IgG4-associated cholangitis,IAC)可能。但因胰周多发曲张静脉,穿刺风险较高,未行胰腺及胆管穿刺取组织学证据。
因无法组织学确诊,只能经验性治疗观察疗效吗?
回顾病史,发现患者尿常规示蛋白微量,余未见异常;24 h尿蛋白0.83 g/24 h↑;血Cr(E)一度高达407 μmol/L;电解质无明显异常。
患者肾功能异常无法用高血压及肾前性因素解释,结合血IgG4明显升高,提示肾脏也可能是受累靶器官。
行肾穿刺检查。
肾穿刺病理:6/12球性硬化,可见节段性系膜细胞增生和系膜基质增生;弥漫性肾小管萎缩,弥漫性间质纤维化,大量炎性细胞浸润 (图4),提示间质性肾炎。免疫组化IgG(+),IgG4(+)>10/HP。
最终诊断:IgG4相关性疾病,IgG4相关性胰腺炎(AIP 1型),IgG4相关性胆管炎,IgG4相关性肾间质病变;高血压病 (2级,高危)。
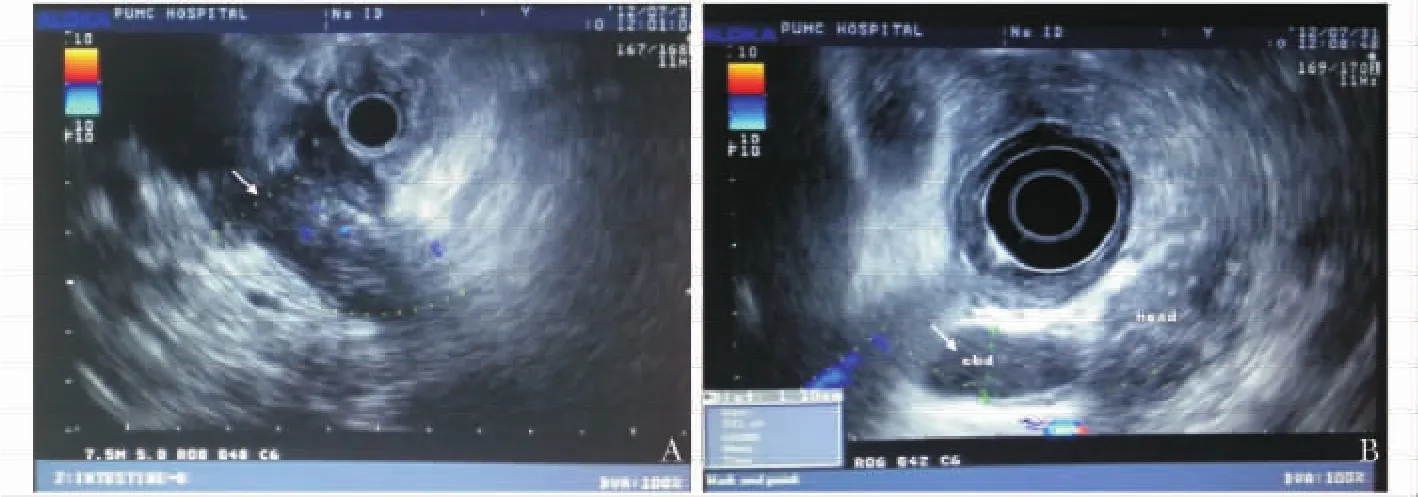
图3内镜下超声检查

图4肾穿刺病理
IgG4-RD,又称IgG4阳性多器官淋巴细胞增生综合征,最早可追朔到1892年Mikulicz[4]发现的1例双侧无痛性泪腺及涎腺肿大疾病,该类疾病最终命名为米库利兹病 (Mikulicz disease,MD),一度被认为是干燥综合征 (Sjgren's syndrome,SS)的亚型。直到2009年,日本学者Masaki和Umehara[5]发现MD肿大的脏器中存在IgG及IgG4阳性细胞浸润,而SS则不存在,才将其明确区分为两类疾病,后归入IgG4-RD疾病谱中。2003年Kamisawa等[6]首次提出IgG4-RD的概念;2010年Autoimmun Rev正式命名IgG4-RD[7]。IgG4-RD发病机制不清,特征性病理改变为多个器官及组织中广泛的IgG4阳性淋巴细胞浸润,可导致相应组织硬化和纤维化。
IgG4-RD的特点:(1)老年人多见,男性为主; (2)病因不清,疾病早期无特异性临床表现;(3)一个或多个器官或组织肿瘤性肿胀,有进行性纤维化,包括胰腺 (AIP)、唾液腺和泪腺 (MD)、肺 (间质性肺炎)、腹膜后间隙 (腹膜后纤维化)、肾 (间质性肾炎)、蛛网膜 (硬脑膜炎)、垂体 (垂体机能减退综合征)等,同时可累及眼眶、肺和乳腺等组织或器官,尤其倾向于导致炎性假瘤,其中以胰腺、唾液腺和泪腺受累最为常见,60%~90% 的患者可同时或先后累及多个器官;(4)IgG4阳性淋巴细胞大量增生导致淋巴细胞增生性浸润和硬化;(5)血清IgG4水平显著升高>1350 mg/L,IgG4阳性淋巴细胞在组织中浸润,IgG4阳性淋巴细胞占淋巴细胞的50%以上; (6)对糖皮质激素治疗反应良好。IgG4-RD诊断流程见图5。
IgG4-RD的治疗:糖皮质激素为首选,起始剂量多为0.6 mg/kg,根据病情2~4周后酌情减量至2.5~5.0 mg/d维持治疗,维持治疗时间尚无统一规定;需注意糖皮质激素治疗的不良反应。酌情使用甲氨蝶呤、咪唑硫嘌呤和霉酚酸酯等免疫抑制剂,以避免或减少糖皮质激素治疗的不良反应及治疗时间。有文献报道,对糖皮质激素抵抗、依赖或复发者,利妥昔单抗 (rituximab)有效。严重胆道梗阻患者,行内镜下胆道支架置入也是较好的选择。

图5IgG4相关性疾病诊断流程图[8]
治疗及转归:在充分知情同意后,患者一方面行补液、保肝、降压等积极对症支持治疗;另一方面根据检查结果,于住院第22天起,予糖皮质激素 (每天泼尼松龙0.6 mg/kg)治疗。患者食欲逐步好转,二便基本正常。糖皮质激素治疗半月后复查肝功能: TBil从469.4 μmol/L恢复至45.3 μmol/L,DBil从352.7 μmol/L恢复至35.9 μmol/L,ALT从218 U/L恢复至45 U/L,ALP从303 U/L恢复至111 U/L,GGT从316 U/L恢复至139 U/L;肾功能:BUN从22.29 mmol/L恢复至8.6 mmol/L,Cr(E)从407 μmol/L恢复至130 μmol/L。ESR从 105 mm/L恢复至 6 mm/h,hsCRP从5.57 mg/L恢复至0.39 mg/L。CT平扫提示肿大的胰腺较入院时 (图1)明显缩小 (图6)。

图6治疗1月后腹部CT平扫,胰腺明显缩小 (箭头)
证实治疗有效。
患者于糖皮质激素治疗半月后出现血糖升高,予胰岛素治疗。首次减量时因一度“嫌麻烦”停服泼尼松,导致严重的继发性皮质功能不全而入院抢救。经多科会诊并及时予氢化可的松25 mg 3次/d及霉酚酸酯250 mg 2次/d治疗 (国外常规2~3 g/d,国内1.5~2 g/d),患者症状明显缓解。复查肝肾功能Alb 37 g/L,TBil 9.9 μmol/L,DBil 4.9 μmol/L,ALT 24 U/L,Cr(E)147 μmol/L;IgG1 6940 mg/L,IgG2 5630 mg/L,IgG3 262 mg/L,IgG4 9700 mg/L。
IgG4-RD为一组新发现的疾病,发病机制尚不明确,诊断标准及治疗方案亦未统一,仍需不断完善相关临床及基础研究。
通过对本病的诊治,笔者的经验如下:(1)老年人群,一般情况好,有不明原因的脏器肿大,在除外恶性疾病后,需考虑IgG4-RD等少见偏良性疾病;注意按上述诊断流程合理诊断,病理是重要的诊断标准,应积极创造条件行相关组织学检查;(2)充分注意并预防糖皮质激素治疗的不良反应,特别对老年人群高血糖、高血压及骨质疏松等较严重的不良反应,要有高度警惕性;(3)加强老年患者的宣教,包括对疾病的正确认识,药物不良反应的风险及预防,规律药物使用的重要性等,最终促成患者认真配合治疗; (4)个体化治疗十分重要。
[1]Yoshida K,Toki F,Takeuchi T,et al.Chronic pancreatitis caused by an autoimmune abnormality:proposal of the concept of autoimmune pancreatitis[J].Dig Dis Sci,1995,40: 1561-1568.
[2]陈刚,卓华,陈国璋.IgG相关硬化性疾病:一种仍在演变中的综合征[J].中华病理学杂志,2010,39:851-868.
[3]韩朝飞,左朝晖,赵佳正,等.自身免疫性胰腺炎的国际统一诊断标准[J].中华胰腺病杂志,2012,12:67-72.
[5]Masaki Y,Umehara H.IgG-related disease-the diagnostic confusion and how to avoid it[J].Jpn J Clin Immunol,2009,32: 478-483.
[6]Kamisawa T,Funata N,Hayashi Y,et al.A new clinicopathological entity of IgG4-related autoimmune disease[J].J Gastroenterol,2003,38:982-984.
[7]Takahashi H,Yamamoto M,Suzuki C,et al.The birthday of a new syndrome:IgG4-related diseases constitute a clinical entity[J].Autoimmun Rev,2010,9:591-594.
[8]Umehara H,Nakajima A,Nakamura T,et al.IgG4-related disease and its pathogenesis-cross-talk between innate and acquired immunity[J].Int Immunol,2014,26:585-595.
R593.9;R442.4;R692.9
A
1674-9081(2015)02-0150-06
10.3969/j.issn.1674-9081.2015.02.015
2014-12-10)
李景南电话:010-69155017,E-mail:lijn2008@126.com
