一种适于丝状真菌基因RNA干扰方法的研究
2015-05-28张振颖侯彬彬刘霞
张振颖 侯彬彬 刘霞
(1.香港大学深圳医院皮肤科,深圳518053;2.大连医科大学附属第二医院皮肤科,大连116023)
RNA干扰(RNAi)是真核生物中相对保守的基因沉默机制,它通过内源或外源双链RNA介导细胞内mRNA发生特异性降解或翻译的抑制,从而导致靶基因的表达沉默,并产生相应的功能表型缺失[1]。1992年,Macino首次在粗糙脉胞菌中发现真菌中存在RNAi的现象后,RNAi技术渐成为一种对真菌进行遗传改造的新手段[2-4]。在丝状真菌中RNAi技术具有特殊的优势:不受丝状真菌的多核现象、异核现象和非同源重组频率高的干扰,特别是当需要使多个基因的表达同时下调时,使用RNA干扰比基因敲除更加方便。然而,真正将RNAi技术用在丝状真菌的各项研究时还存在很多问题:真菌细胞多为多倍体,细胞壁含有较多的多糖及多糖蛋白等成分,使得RNA沉默的效果并不如在原生、动物细胞中理想,存在干扰效率低下、有脱靶效应等缺陷。因此选择恰当的遗传转化方法并制备合适的转染载体,可以更高效地导入siRNA,保证导入siRNA的稳定性,有效延长RNAi的作用时间,进而大大提高靶基因抑制效率,更好地对丝状真菌进行基因改造。
1 材料与方法
1.1 材料
菌株 DH5α,申克孢子丝菌ATCC10268。
质粒 pBI121,pBlueScriptII,pSilent-1(the Fungal Genetics Stock Center)。
酶和主要试剂 XbaI、XhoI、SpeI、SacI、KpnI、BglII、HindIII等限制性内切酶,T4 DNA连接酶,Taq酶、T载体。DNA分子量Marker(Marker III)、质粒提取试剂盒、DNA纯化回收试剂盒。
培养基 ①LB(Luria-Bertani)液体培养基(g/L)。②LB(Luria-Bertani)固体培养基 (g/L)。③诱导培养基(IM):在蒸馏水中溶解15 g琼脂至900.7 mL,高压后加入 0.8 mL K-buffer,20 mL MN-buffer,1 mL 1%CaCl2·2H2O,10 mL 0.01%FeSO4,5 mL 微量元素,2.5 mL 20%NH4NO3,10 mL 50%甘油,40 mL pH5.5 的 1 mmol/L MES,10 mL 的 20%葡萄糖。④筛选培养基 (SM):IM培养基中加入100μg/mL潮霉素,0.2 mol/L头孢噻肟钠。
1.2 重组载体PCB309-pfgrt的构建
感受态细胞的制备 将DH5α菌保接到LB液体培养基中,37℃,220 r/min,培养16 h。然后转接到50 mL LB液体培养基,220 r/min,37℃培养到OD600 0.6。将培养物冰上预冷 10 min,4℃,4 000 r/min离心5 min,弃上清。用预冷30 mL的CaC12(0.1 mol/L)重悬,冰上放置 30 min,4℃,4 000 r/min离心 5 min,弃上清。最后用 1 mL预冷的CaC12(0.1 mol/L)重悬,用枪吹吸,使其均匀,按100 μL/管分装。
转化感受态细胞 每管感受态细胞加入连接产物10μL,轻混匀,冰上放置30 min,42℃的水浴中,热激70 s。快速转移到冰浴中,冷却2 min。每管加入900μL的液体LB,37℃摇床温育60 min,使细菌复苏并表达质粒编码的抗生素标记抗性基因。4 000 r/min离心后用100μL液体LB重悬,涂布于含有相应抗生素的固体LB平板上。倒置于37℃培养,16 h后挑选阳性克隆,提取质粒,酶切验证。
PCB309的构建 ①构建PCB298:以pBI121为模板,用引物PCB298-F和PCB298-R扩增RK2复制区和 nptⅡ,长 4 566 bp(94℃ 1 min;94℃ 30 s,55℃45 s,72℃ 4 min,30 个循环,72℃ 10 min)。PCR 片段纯化回收,用XbaI单酶切,酶切后再纯化回收。酶切后的纯化片段进行连接,将单片段连接为环形质粒。连接产物转化到大肠杆菌DH5α,挑取单菌落,提质粒,XbaI单酶切验证。②构建PCB299:以PCB298为模板,用引物PCB299-F和PCB299-R扩增RK2复制区和nptⅡ去除is1非必须区,得到片段长度 3 166 bp(94℃ 1 min;94℃ 30 s,55℃ 45 s,72℃ 2 min,30个循环72℃ 10 min)。将PCR片段纯化回收,用XhoI单酶切,酶切后再纯化回收。酶切后的纯化片段进行连接,将单片段连接为环形质粒。将连接产物转化到大肠杆菌DH5α,挑取单菌落,提质粒,用XhoI酶切验证。③构建PCB300:以pBI121为模板,用引物PCB300-F和PCB300-R扩增pBI121的T-DNA全部区域,得到片段长度6 373 bp(94℃ 1 min;94℃ 30 s,55℃ 45 s,72℃ 6 min,30 个循环,72℃ 10 min)。将PCR片段纯化回收,用SpeI单酶切;将PCB299用XbaI单酶切。酶切后再分别纯化回收。酶切后的纯化PCR片段与PCB299进行连接,连接为环形质粒。将连接产物转化到大肠杆菌DH5α,挑取单菌落,提质粒,用引物PCB298-F和PCB298-R鉴定。④构建PCB301:以PCB300为模板,用引物PCB301-F和PCB301-R扩增T-DNA边界序列和PCB300的序列,得到片段长度3 400 bp(94℃ 1 min;94℃ 30 s,55℃ 45 s,72℃ 3 min,30 个循环,72℃ 10 min)。将PCR片段纯化回收,用Sac I和KpnI双酶切;将pBlueScriptII用Sac I和KpnI双酶切。酶切后再分别纯化回收。酶切后的纯化PCR片段与pBlueScriptII切下来的多克隆位点(MCS)进行连接,连接为环形质粒得到PCB301。将连接产物转化到大肠杆菌DH5α,挑取单菌落,提质粒,测序验证。⑤构建PCB309:以pSilent-1质粒为模板,用引物HPH-F和HPH-R扩增Ptrpc启动子、潮霉素抗性基因和Ttrpc终止子的序列,得到片段长度约1.8 kp(94℃ 1 min;94℃ 30 s,55℃ 45 s,72℃ 2 min,30 个循环,72℃ 10 min)。将PCR片段纯化回收,用KpnI和HindIII双酶切;将PCB301用KpnI和HindIII双酶切。酶切后再分别纯化回收。将酶切后的纯化PCR片段与酶切后纯化的PCB301载体进行连接,连接为环形质粒得到PCB309。将连接产物转化到大肠杆菌DH5α,挑取单菌落,提质粒,KpnI和HindIII双酶切验证。
RNA干扰载体PCB309-pfgrt的构建 ①构建PUC-pgt:以pSilent-1质粒为模板,用引物PUT-F和PUT-R扩增Ptrpc启动子、间隔序列和Ttrpc终止子的序列,得到片段长度约2.2 kp(94℃ 1 min;94℃ 30 s,55℃ 45 s,72℃ 3 min,30 个循环,72℃10 min)。将纯化后的PCR片段与Simple T载体进行连接,连接为环形质粒得到PUC-PUT。将连接产物转化到大肠杆菌DH5α,挑取单菌落,提质粒,用引物PUT-F和PUT-R进行PCR验证。②构建PUC-pfgrt:扩增靶基因正向干扰序列,纯化回收产物经酶切后与PUC-pgt载体连接为环形质粒得到PUC-pfgt;扩增靶基因反向干扰序列,纯化回收产物经酶切后与PUC-pfgt载体连接为环形质粒得到PUC-pfgrt。③实验所用引物见表1。④构建PCB309-pfgrt:将PUC-pfgrt和 PCB309分别用 SpeI和SacI双酶切。酶切后再分别纯化回收 (37℃,3 h)。再进行连接,连接为环形质粒得到PCB309-pfgrt(22℃,2 h)。⑤将连接产物转化到大肠杆菌DH5α,挑取单菌落,提质粒,SpeI和SacI双酶切酶切验证。

表1 文中所用引物序列列表Tab.1 Primers sequences used in this experiment
根癌农杆菌感受态细胞的制备 ①由冰箱保存的菌种EHA105划板以备挑单菌落SOB或LB固体平板,不加抗生素,常规3~4区划板。②接种感受态细胞:挑一个单菌落1~2 mL SOB液体培养基中(利福平50μg/mL)。③将接菌后的试管于28℃,300 r/min,培养 6 h,导入 20 mL SOB 液体培养基中,300 r/min,37℃培养到 3 h时,测 1次OD600。④之后每10 min测1次,当OD值接近0.76时,每2 min测1次,至 OD=0.76时,将菌液置冰水浴中,不断摇动,随后在50 mL离心管(预冷)中离心,4℃,2 000 g,10 min,弃上清。用预冷的灭菌重蒸水洗,离心2 200 g,10 min,弃上清。⑤再用预冷的10%甘油(V/V)以同样方法洗两次,离心2 400 g,10 min,最后1 次倒尽甘油,分装,用-80℃的乙醇迅速冰冻,-80℃保存备用。
质粒PCB309-pfgrt转化根癌农杆菌感受态细胞,挑选阳性克隆验证 ①将质粒采用电击的方法转化根癌农杆菌,冰上操作,取待转化的质粒PCB309-pfgrt和PCB309各1μL混入感受态细胞中,混匀。②吸取置于已经预冷的电击杯中,擦去电击杯外的冷凝水和蒸汽,推入电击仪中,电压为1.8 V,在电击杯中加入1 mL LB培养基,冲洗,使转化后的细胞与培养基充分混合,吸取入试管中,28℃,1 h。③将培养好的菌液加入EP管中离心,弃上清,用剩余的液体重悬菌体,全部涂到含有抗生素Kan的LB培养基中 (Kan 50 mg/mL,利福平25 mg/mL)。④培养2 d后挑取阳性克隆,提取质粒,验证。用引物HPH-F和HPH-R扩增Ptrpc启动子、潮霉素抗性基因和Ttrpc终止子的序列,得到片段长度约 1.8 kp(94℃ 1 min;94℃ 30 s,55℃ 45 s,72℃ 2 min,30 个循环,72℃ 10 min)。
根癌农杆菌介导重组载体PCB309-pfgrt对孢子丝菌DRK1基因进行干扰 ①确定孢子丝菌对潮霉素的敏感性:将孢子丝菌分生孢子 (1.0×104个/mL,涂100μL)接种于含有不同浓度潮霉素的沙氏培养基上 (25℃,5 d),确定最佳使用浓度为100μg/mL。②a.分生孢子的制备:将分生孢子接种于沙氏培养基,25℃,5 d;b.在已产孢的沙氏培养基,加入1 mL水,轻轻震荡,收集分生孢子;c.使用血细胞计数板确定孢子浓度,达到最终浓度为5.0×106个/mL。③农杆菌细胞的制备:a.将农杆菌菌保接种于含有20μg/mL Rif和100μg/mL Kan的LB 液基中,28℃ 24 h,200 r/min;b.将上述培养农杆菌细胞收集于1.5 mL离心管,室温,2 400 g离心10 min,弃上清;c.用IM液体重悬细胞,室温,2 400 g离心10 min,弃上清;d.在100 mL锥形瓶中,将农杆菌细胞重悬于15 mL添加200μmol/L AS的IM培养基,28℃ 8 h,100 r/min;e.当 OD 600 nm 为 0~0.8时,用于转化。④农杆菌与孢子丝菌共培养:a.取1 mL OD 600 nm 为 0.6~0.8 的农杆菌细胞与1mL浓度为5.0×106个/mL孢子丝菌分生孢子混合;b.将上述200μL混合液均匀涂布于1 M+200 μmol/AS和IM-AS的平板上,25℃避光培养。筛选转化子:将共培养混合物的转移到诱导平板上 (含100μg/mL潮霉素、0.2 mol/L头孢噻肟钠)25℃ 4 h,直至菌落出现;将具有潮霉素抗性的孢子丝菌单菌落转接于沙氏培养基,4℃保存。⑤检测转化子的遗传稳定性:随机挑选不同批次的转化实验的20个转化子,在无潮霉素的沙氏培养基上连续传代5次后,转移至含有100 mg/L潮霉素的SM平板上,观察转化子是否仍具有潮霉素抗性,检测转化子的遗传稳定性。
1.3 干扰效率的检测
应用Real-Time PCR方法对申克孢子丝菌DRK1组氨酸激酶基因mRNA相对表达量进行分析;应用SYBR GreenⅠ荧光染料嵌合法,以1#样品Total RNA反转录获得的cDNA作为标准品,分别对管家基因(18S)和目的基因 (DRK1)制作标准曲线;然后再分别对各样品的管家基因和目的基因进行定量实验,通过管家基因的校正,对各样品中目的基因的相对表达量进行分析。
实验试剂及引物 ①TaKaRa RNAiso Plus Reagent。②PrimeScript RT reagent Kit with gDNA E-raser。③SYBR Premix Ex Taq(Tli RNaseH Plus);18S-F(5’-agcggagggatcattacagag-3’);18S-R(5’-cgccagaagcaacgagaa-3’);DRK1-F(5’-CGATGAGTACGCGATTGAGTTT-3’);DRK1-R(5’-CGCTTGGAAATGGAAAGACC-3’)。
实验步骤 ①总RNA质量的确认,OD值测量。②Real Time PCR法检测目的基因相对表达量。
反应组成及反应条件 ①基因组DNA去除反应 (5×gDNA Eraser Buffer 2μL;gDNA Eraser 1μL;Total RNA;1μL;RNase Free dH2O 6μL;42℃ 2 min)。②RT反应 (上述反应液10μL;5×Primer-Script Buffer 2(for Real Time)4μL;PrimeScript RT Enzyme Mix I 1μL;RT Primer mix 1μL;RNase Free dH2O 4μL;37℃ 15 min 85℃ 5 s)。③PCR反应(SYBR Premix Ex Taq II(Tli RNaseH Plus)(2×)12.5 μL;Primer F(10 μmol/L)1 μL;Primer R(10 μmol/L)1 μL;RT产物2μL;dH2O 8.5μL;95℃ 30 s,95℃ 5 s,60℃ 30 s,40Cycles)。
2 结 果
2.1 干扰载体构建过程中涉及到的酶切及PCR鉴定图 (见图1~2)
2.2 应用Real-Time PCR方法进行申克孢子丝菌组氨酸激酶DRK1基因相对表达量分析
我们利用Real-Time PCR方法对申克孢子丝菌组氨酸激酶DRK1基因相对表达量分析,从结果可见干扰后组氨酸激酶DRK1基因表达水平发生变化,以37℃野生标准株DRK1相对表达量为标准1,干扰后37℃干扰株DRK1相对表达量为0.11;25℃野生标准株DRK1相对表达量为0.41,干扰后25℃干扰株DRK1相对表达量为0.12。与菌丝相相比较,酵母相DRK1的表达水平明显升高,干扰后组氨酸激酶DRK1基因表达明显降低(见图3)。
3 讨 论
目前存在的遗传转化方法众多:CaCl2/聚乙二醇法,醋酸锂转化法,电穿孔转化法,粒子轰击法和根癌农杆菌转化法。与其他方法相比,在丝状真菌的研究中根癌农杆菌转化法具有如下优势:①不需制备原生质体,各种类型的完整细胞 (如分生孢子、菌丝体、子实体)均可作为转化受体。②产生的突变子大多数为单拷贝插入,其标签基因的分离更容易。③插入随机性更好,亦能用于同源重组。由此可见,根癌农杆菌介导的真菌遗传转化方法不失为真菌遗传学研究领域的一种重要手段,而构建适于农杆菌介导的、能够转染丝状真菌的干扰载体尤为必要[5-10]。常用的植物双元表达载体都比较大,多数在10 kb以上。做载体构建时操作困难,转化效率低,而且可用的单一酶切位点很少。本研究对现有的植物双元表达载体进行改造,而改造后的载体只有3.5 Kb,并加入了多克隆位点,从而适于真菌基因的干扰沉默研究中。另外,目前文献报道干扰载体的适用范围多较局限,为种属特异性,仅适于皮肤癣菌属/青霉菌属/曲霉菌属,大大限制了其应用范围。我们将改造后的载体加入了真菌通用启动子Ptrpc、间隔序列和终止子Ttrpc,并引入了潮霉素抗性基因,使其在间隔序列两侧加上目的基因的正反向干扰序列后即可广泛用在多种真菌基因干扰的研究中。现存的很多真菌基因干扰方法存在转化效率低,稳定性差等缺点,本研究使用根癌农杆菌的转化体系,并对转化体系中诱导剂的种类和浓度、培养时间、培养温度、根癌农杆菌与受体菌浓度之比等条件参数进行优化,提高了转化效率,保证了导入siRNA的稳定性,明显提高了靶基因的抑制效率。
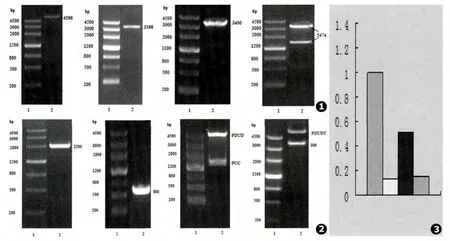
图1 PCB298酶切鉴定图,PCB299酶切鉴定图,PCB300 PCR鉴定图,PCB309酶切鉴定图 图2 PUC-PUT PCR鉴定图,PUC-Pfgt PCR鉴定图,PUC-pfgrt酶切鉴定图,PCB309-pfgrtT酶切鉴定图 图3 干扰前后孢子丝菌菌株的DRK1mRNA表达水平的变化。依次为:37℃野生标准株;37℃干扰株;25℃野生标准株;25℃干扰株Fig.1 PCB298 restriction map,PCB299 restriction map,PCB300 PCR product,PCB309 restriction map Fig.2 PUC-PUT PCR product,PUC-Pfgt PCR product,PUC-Pfgrt restriction map,PCB309-Pfgrt restriction map Fig.3 mRNA expression levels of DRK1 in Sprothrix schenckii before and after interference:wild type strain at 37℃,interference strain at 37℃,wild type strain at 25℃,interference strain at 25℃
本研究提供了一种由根癌农杆菌介导的适于丝状真菌基因研究用的RNA干扰方法,具体如下:干扰载体构建,包括干扰载体的序列特征,目的基因正反相干扰序列替换的位置,载体构建的具体条件;根癌农杆菌转化体系中供体菌与受体菌浓度之比,培养温度,培养时间,诱导剂种类和浓度等条件参数。我们应用该体系实现了对孢子丝菌双组份信号传导通路中组氨酸蛋白激酶DRK1基因的有效修饰,明显下调了该基因的表达水平,干扰后其表达水平仅为野生株11%。
[1] Hammond SM,Caudy AA,Hannon GJ.Post-transcriptional gene silencing by double-stranded RNA[J].Nat Rev Genet,2001,2(2):110-119.
[2] Chad A.Rappleye,Jacquelyn T,et al.RNA interference in Histoplasma capsulatum demonstrates a role for a-(1,3)-glucan in virule[J].Mol Microbiol,2004,53(1):153-165.
[3] Sommer U,Liu H,Doering TL.An alohal,3-mannosyltrans-ferase of Cryptococcus neoformans[J].Bioi Chem,2003,278(48):47724-47730.
[4] Isabelle Mouynaa,Christine Hemya,Tamara L,et al.Gene silencing with RNA interference in the hunm pathogenic fungus Aspergillus fumigatus[J].FEMS Microbiol Lett,2004,237(2):317-324.
[5] Fincham JRS.Transformation in fungi[J].Microbiol Review,1989,53(1):148-170.
[6] Dos Reis MC,Pelegrinelli Fungaro MH,Delgado Duarte RT,et al.Agrobacterium tumefaciens-mediated genetic transformation of the entomopathogenic fungus Beauveria bassiana[J],2004,58(2):197-202.
[7] Mullins ED,Chen X,Romaine P,et al.Agrobacterium-mediated transformation of Fusarium oxysporum:an efficient tool for insertional mutagenesis and gene transfer[J].Phytopathology,2001,91(2):173-180.
[8] Gento Tsuji,Satoshi Fujii,Naoki Fujihara,et al.Agrobacterium tumefaciens-mediated transformation for random insertional mutagenesis in Colletotrichum lagenarium [J].Journal of General Plant Pathology,August 2003,69,(4):230-239.
[9] Dobinson KF1,Grant SJ,Kang S.Cloning and targeted disruption,via Agrobacterium tumefaciens-mediated transformation,of a trypsin protease gene from the vascular wilt fungus Verticillium dahliae[J].Curr Genet,2004,45(2):104-110.
[10] De Groot MJ1,Bundock P,Hooykaas PJ,et al.Agrobacterium tumefaciens-mediated transformation of filamentous fungi[J].Nat Biotechnol,1998,16(9):839-842.
