Unusual Cardiomyopathies: Some May Be More Usual Than Previously Thought and Simply Underdiagnosed
2015-05-22FrankSmartMDFACCFACP
Frank W. Smart, MD, FACC, FACP
1Louisiana State University Health Sciences Center, New Orleans, LA, USA
Introduction
Heart failure is the most common cause of hospitalization in the Medicare age population. Both systolic and diastolic abnormalities of cardiac performance result in the heart failure syndrome. The associated cardiomyopathies resulting in clinical heart failure are still being def i ned with regard to their incidence and prevalence.
Unusual cardiomyopathies are typically characterized as such because they are believed to occur very infrequently, and represent a small portion of those individuals with nonischemic cardiomyopathy. This def i nition carries with it a geographic-specific implication. That is, cardiomyopathies around the world differ in their incidence and prevalence.Chagas disease, for example, would be unusual in the Midwestern United States, but would be quite usual in heart failure clinics in South America.
Many unusual cardiomyopathies have been seen with increasing frequency in the United States.This increase is likely due to improved imaging techniques, and increased sensitivity of the heart failure cardiologist to the true prevalence of these cardiomyopathies. Two such causes of heart failure are isolated left ventricular noncompaction (LVNC)and cardiac amyloidosis. Both of these cardiomyopathies have been associated with various genetic abnormalities, but because genetic testing is still prohibitively expensive, genetic tests are not routinely performed on individuals who present with congestive heart failure. Perhaps in the future, genetic testing will be incorporated to better characterize first the diagnosis and second the prognosis that is associated with these illnesses. Although standard treatments for congestive heart failure should be applied to both LVNC and cardiac amyloidosis, certain nuances in the diagnosis of LVNC and cardiac amyloidosis and in the treatment of individuals with LVNC or cardiac amyloidosis require further characterization. A keen awareness of the specifics of the pathophysiologic state is essential in both entities since patients can remain phenotypically silent and only manifest decompensation in the presence of another physiologic or environmental stressor.
The purpose of this review is to offer insight into the incidence and prevalence of the unusual cardiomyopathies, which may be much more usual than previously thought. In addition, the aim is to focus on the differences between these specific causes of congestive heart failure and the more common causes of both heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF).
Left Ventricular Noncompaction
Initially described as a pathologic finding seen in congenital heart defects, LVNC was characterized as abnormal coronary distribution by Grant [1] in the early 1900s. The presence of thickened myocardium with multiple trabeculations and deep clefts was known to congenital heart disease specialists as a rare association of the myocardium that resulted from the arrest of maturation of the fetal heart tube [2].
The fetal heart tube is the first stage of development of the cardiovascular system in utero. As the fetus grows and matures, the heart develops multiple trabeculations, which signifi cantly increase the cardiac mass without requiring epicardial coronary fl ow as the primary source for myocardial oxygen and nutrients. At about 8 weeks of gestation, trabecular remodeling begins to occur. In this stage the trabeculations began to coalesce to form normal endocardial structures, including the papillary muscles.
As coronary arteries develop through angiogenesis, further remodeling of the myocardium occurs such that a two-layered ventricular wall is formed.The first layer is the compacted layer with a spiral arrangement of ventricular myocytes; and the second, noncompacted or trabeculated layer, has more random myocyte organization and subsequently generates less organized contractile force. Compaction occurs from epicardium to endocardium and from the base to the apex of the heart. An arrest in the compaction process for any reason will therefore be variable among individuals, but LVNC will always involve the endocardial surface of the apex of the left ventricle since this is the last area to undergo compaction [3]. The ratio of the compacted to noncompacted layers of the heart governs the performance characteristic of the adult heart and is one of the diagnostic criteria used to characterize LVNC [4]. When the noncompacted layer of the myocardium is excessive and occurs in the absence of any other congenital cardiac abnormality, the patient is said to haveisolatedLVNC.
There have been many hypotheses as to why the ventricular myocardial remodeling process is arrested. LVNC is associated with 15 abnormal genes, including those coding for sarcomeric, mitochondrial, cytoskeletal, and Z line proteins [5].LVNC patterns of inheritance that have been noted include X-linked, autosomal dominant, autosomal recessive, and mitochondrial patterns. The most frequently identif i ed gene abnormalities are those of sarcomeric protein genes coding for β-myosin heavy chain and α-actin. There is an undulating phenotype in LVNC such that affected individuals can suddenly become worse with little warning [6].Initially it was thought that LVNC affected 0.12 in 100,000 individuals [7]. This prevalence was similar to what was described in hypertrophic cardiomyopathy when it was initially characterized. Today,with increasing imaging capability and genetic screening,hypertrophic cardiomyopathyis thought to affect one in 500 individuals. LVNC has very similar genetics, and may well be on the spectrum between dilated cardiomyopathy and hypertrophic cardiomyopathy [8]. Investigators today believe that LVNC may account for as many as 4%–5% of all individuals with HFrEF [8]. Considering dilated cardiomyopathy accounts for 40% of all HFrEF,LVNC is not such an uncommon diagnosis.
Some X-linked cardiomyopathies that resemble LVNC, such as Barth syndrome,exist in children but have not been seen in the adult population because of the limited survival of individuals with X-linked cardiomyopathies [9].
Epigenetic changes in DNA methylation, chromatin remodeling, or histone posttranslational modif i cation have also been implicated in the development of LVNC. Some maternal conditions that would result in fetal hypoxia have been shown in animal models to cause such epigenetic changes [10]. Similar changes have also been noted with maternal use of cocaine. Some controversy is emerging as to whether LVNC can be acquired in the adult. Adults who meet the diagnostic criteria by echocardiography for LVNC may have no heart failure signs or symptoms at all. These individuals appear to have the LVNC genotype but as yet have not phenotypically manifested the classic LVNC characteristics. Other investigators propose these affected individuals have enough compacted myocardium to remain asymptomatic, but another environmental or morphologic insult can result in heart failure from LVNC simply by increasing the demand on myocardial performance, or by further expressing epigenetic alterations to contractile or ultrastructural proteins [8].
Myocardial ischemia may also have a profound effect in converting someone with asymptomatic LVNC to someone with HFrEF [6]. The arrested coronary artery development and the susceptibility of large trabeculations to myocardial ischemia from capillary collapse as a result of increased left ventricular end-diastolic pressure can begin the cycle of impaired cardiac performance leading to increased left ventricular end-diastolic pressure, which in turn causes more ischemia and then more heart failure.Epicardial coronary lesions in patients will hasten the process.
Failure of the ventricular myocardium to further compact alters the contraction properties of the heart in addition to the morphologic appearance on echocardiography and left ventriculography.Diagnosis of this condition is challenging since a slightly off-axis echo resembles LVNC [4]. The altered organization of a larger proportion of ventricular myocytes results in an abnormal twisting of the left ventricle during systole and therefore is more easily identif ied by echocardiographic imaging techniques such as tissue Doppler imaging with speckled tracking and strain rate [11]. These new echocardiographic fi ndings help to provide further differentiation between true LVNC and a hypertrabeculated heart. Many echocardiographers unfortunately fail to routinely use these diagnostic tools when evaluating a patient with HFrEF.
Diagnosis of LVNC
Patients with LVNC usually present with congestive heart failure; however, ventricular tachycardia, sudden cardiovascular death, and systemic embolic events are not uncommon presentations for patients with noncompaction [6]. As many as 10%of patients may be identif i ed on pathologic examination of an explanted heart at the time of cardiac transplantation.
Familial manifestation occurs in roughly 25%of patients, so an in-depth family history is essential when one is evaluating patients with HFrEF.Although sorting out a family history is a seemingly easy task, many individuals believe a family member died of a “heart attack” rather than congestive heart failure or sudden cardiac death. Specif i c mention of an enlarged heart or circumstances surrounding a sudden cardiac death may provide very useful clues to the identif i cation of a familial manifestation of LVNC. Current recommendations state that first-degree relatives of patients with LVNC should be screened with echocardiography [11].
Echocardiography continues to be the mainstay of diagnosis of noncompaction [12]. However, technical challenges result in multiple echocardiograms being performed before the diagnosis is made.Often confused with an idiopathic dilated cardiomyopathy, LVNC has a visible noncompacted layer that is at least two times as thick as the compacted layer [4]. The first challenge is to differentiate an echocardiogram that is obtained in a slightly offaxis fashion and appears hypertrabeculated from a properly obtained echocardiogram showing LVNC.The next major challenge occurs as the sonographer can make adjustments during the echocardiogram acquisition to show less of the spongy endocardial surface, resulting in an echocardiogram that looks more like that corresponding to conventional dilated cardiomyopathy (Figures 1 and 2).
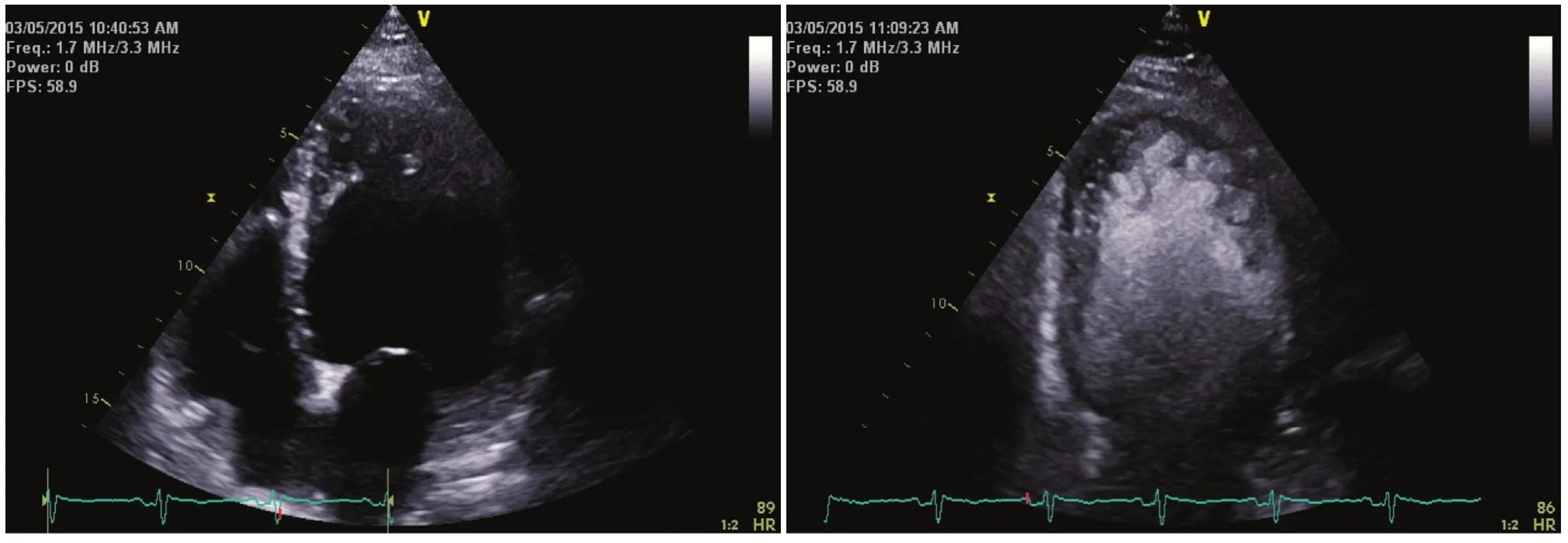
Figure 1 The Left Panel Shows an Apical Four-chamber View of a Patient with Dilated Cardiomyopathy.
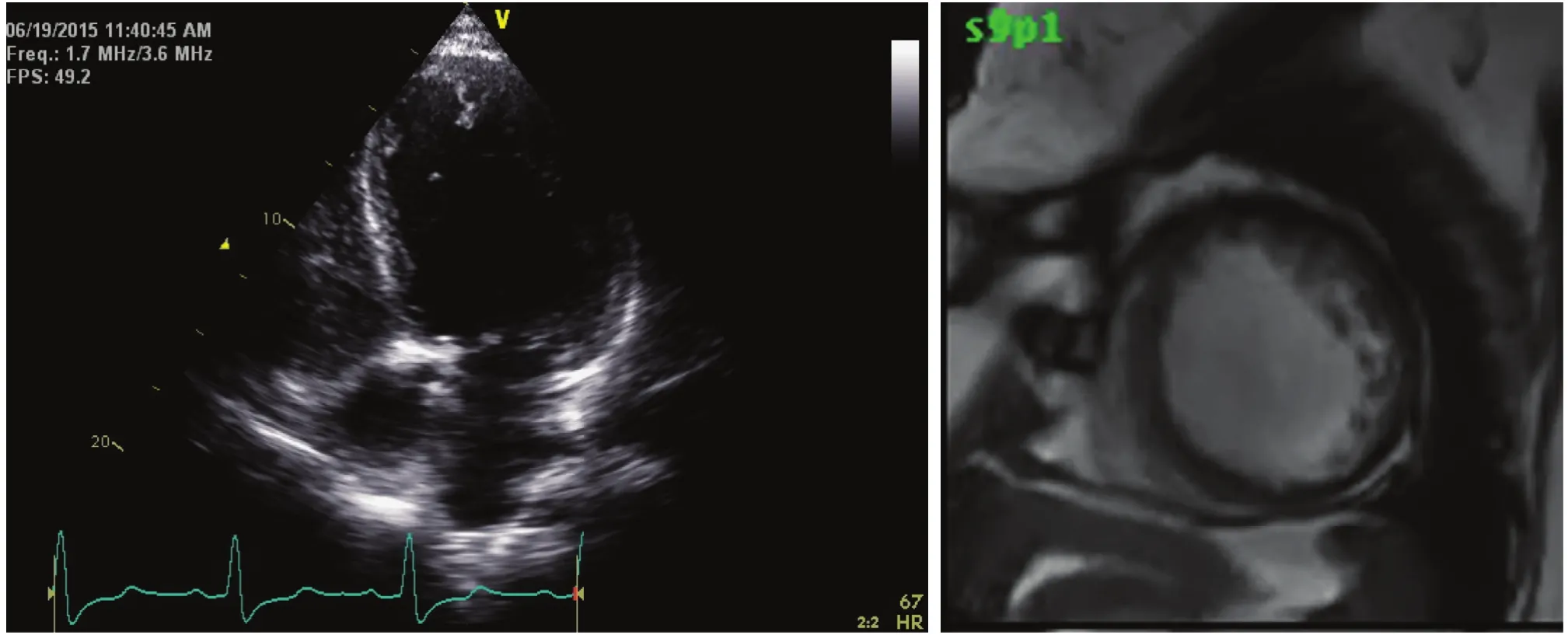
Figure 2 The Left Panel Shows an Apical Four-chamber View of an Echocardiogram with an Inappropriately Low Gain Setting, which was Interpreted as Showing Dilated Cardiomyopathy.
In addition, the use of tissue Doppler imaging,speckle tracking, and longitudinal and circumferential strain provides valuable insight into whether a heart is simply hypertrabeculated or is affected with LVNC [9].
In any echocardiographic study where LVNC is suspected, prudent use of intravenous contrast medium is very helpful in outlining the true endocardial surface, and this technique is well suited for showing deep clefts that occur in the noncompacted layer [12]. The presence of clefts all from the endocardial surface to the compacted layer is one of the criteria used to diagnose LVNC. Table 1 presents a modif i cation of the criteria of Jenni et al. [4] for the echocardiographic diagnosis of isolated LVNC.
Cardiac magnetic resonance imaging (CMRI)has emerged as the diagnostic modality of choice for LVNC [13]. This is because CMRI has signif i -cantly increased contrast and improved spatial resolution when compared with echocardiography. As with echocardiography, the determination of two distinct layers of myocardium is important. Thespongiform layer should be 2.3 times the thickness of the compacted layer in diastole. Again, the apex and lateral walls are the most frequently involved areas. Delayed hyperenhancement of the myocardial trabeculae has also been associated with a more advanced stage of clinical decompensation in patients with LVNC [14].
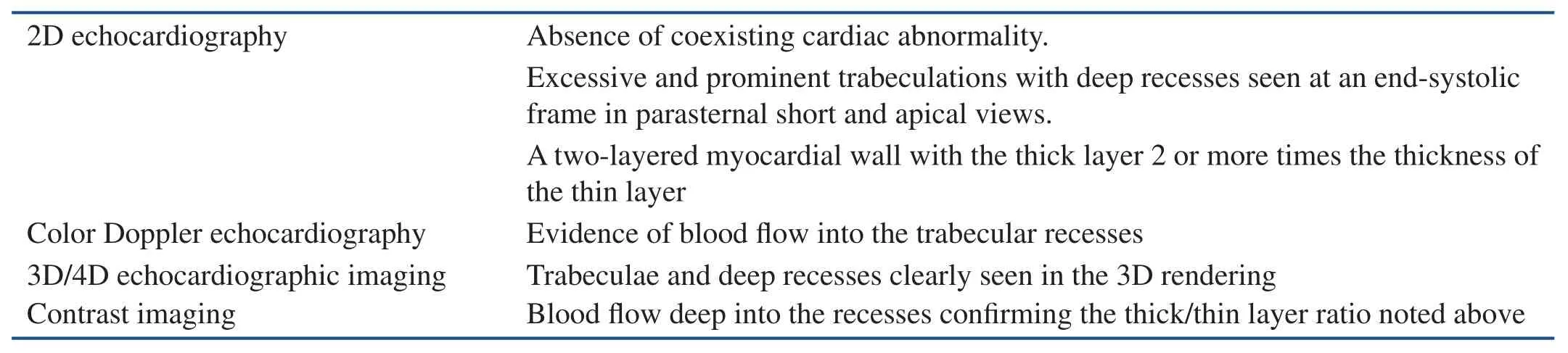
Table 1 Echocardiographic Diagnosis of LVNC.
More recently, cardiac CT has also been described as a sensitive and specific tool in the diagnosis of LVNC. A noncompacted layer greater than 2.3 times the diameter of the compacted layer in diastole seen on CT angiography is diagnostic of LVNC with a similar predictive value to CMRI [15]. Since the use of cardiac CT angiography continues to gain popularity as an imaging modality to def i ne the coronary anatomy in dilated cardiomyopathy, protocols to also visualize the left ventricular cavity to exclude LVNC are currently being ref i ned.
LVNC Treatment
The treatment of LVNC that has resulted in HFrEF is similar to that of other cardiomyopathies, with beta blockers and ACE inhibitors as the mainstay of therapy. Because of the intratrabecular recesses,systemic anticoagulation should be considered in individuals who have a signifi cantly reduced ejection fraction related to LVNC [16].
The prognosis for recovery of patients with LVNC is less robust than that of the general population with dilated cardiomyopathy. Patients who have progressed to advanced stages of heart failure should be monitored closely and considered for advanced heart failure treatments such as cardiac transplantation and ventricular assist systems if they manifest signs and symptoms of end-organ dysfunction, or if the physician needs to reduce ACE inhibitor or beta blocker therapy because of hypotension.
Infiltrative Cardiomyopathy(Amyloidosis)
Infiltrative cardiomyopathies, in general, represent a group of diseases that are characterized by restrictive cardiac physiology. Often the ejection fraction is preserved yet patients have severe limitations in their exercise tolerance. As with most restrictive heart disease, any increase in heart rate will signif i -cantly worsen the heart failure state. Infiltrative cardiomyopathy is one of the severest forms of HFpEF.Cardiac amyloidosis can result from multiple different pathologic abnormalities, and the severity and time course of the disease are quite variable among these types [17].
Immunoglobulin amyloidosis is the first type and is characterized by systemic overproduction of immunoglobulin light chain protein. The conditions commonly associated with this overproduction are plasma cell dyscrasias, Waldenstrom macroglobulinemia, and multiple myeloma [18].
Familial amyloidosis is a genetic abnormality associated with the transthyretin gene. There have been more than 70 different mutations of the genes coding for transthyretin, and the defect is transmitted in an autosomal dominant fashion [19]. Other proteins also can be affected by these mutations and can contribute to the heterogeneity of familial amyloidosis [18].
Senile systemic amyloidosis usually involves the atria of the heart, and patients often exhibit very few symptoms. It has been recognized recently, however, that signif i cant heart failure symptoms can result from senile amyloidosis, and the disease pro-cess can persist for many years [20]. Recent reviews suggest senile amyloidosis is far more common than initially thought. The disease may be present in up to 80% of 80-year-olds [20], and may result in symptoms in 25% of people older than 80 years[20, 21]. Atrial fi brillation and other conduction abnormalities are also seen in patients with senile amyloidosis [22].
Secondary amyloidosis is a condition resulting from systemic inf l ammation from other disease types such as rheumatoid arthritis, chronic lung disease, inf l ammatory bowel disease, and other diseases associated with chronic inf l ammation. Longterm overproduction of acute-phase reactants in these syndromes results in amyloid protein deposition in the heart, with the advanced stages having signif i cant restrictive cardiomyopathy [18].
Hemodialysis-associated amyloidosis results from β2-microglobulin deposition in patients who are receiving long-term hemodialysis as renal replacement therapy. This protein deposition is usually seen in bones and joints; however, evidence is emerging that amyloid protein deposition does occur in the heart [23, 24] and over years could be responsible for heart failure associated with this type of systemic amyloidosis.
Cardiac Amyloidosis Diagnosis
Endomyocardial biopsy has been the gold standard for the diagnosis of cardiac amyloidosis [17]. It is not necessary to perform a heart biopsy when the clinical and echocardiographic fi ndings are present in an individual with biopsy-proven amyloid from another tissue. Subcutaneous fat pad biopsy has a sensitivity of 70% for detecting systemic amyloid [25]. The search for a noninvasive diagnostic modality for cardiac amyloidosis has improved dramatically with the emergence of CMRI; this noninvasive modality has taken a def i nitive position as the noninvasive diagnostic modality of choice for amyloidosis of the heart [26]. The specificity of CMRI is quite high; the current limitation is the sensitivity of detecting the amyloid inf i ltrates when they are small with less clinical impact.
Two-dimensional echocardiography remains a first-line diagnostic tool because of the ubiquitous use of this modality in patients with heart failure symptoms [22, 27]. Like other forms of HFpEF,amyloidosis is associated with impaired filling seen with standard and tissue Doppler assessments. The glistening or speckle pattern noted in the left ventricular walls and extensive atrial enlargement should raise concern for the presence of amyloidosis. In the presence of signif i cant left ventricular hypertrophy on such an echocardiogram, an ECG with low voltage in the limb leads is very specific for the presence of cardiac amyloidosis (Figure 3) [28].
Treatment of cardiac amyloidosis remains very challenging and focuses primarily on the treatment of any underlying disease state resulting in amyloid protein deposition. Numerous clinical trials are currently ongoing looking for pharmacologic agents that will reduce amyloid protein deposition. Cardiac transplantation remains controversial in systemic amyloidosis because of the reoccurrence of amyloid protein deposition in the transplanted heart.
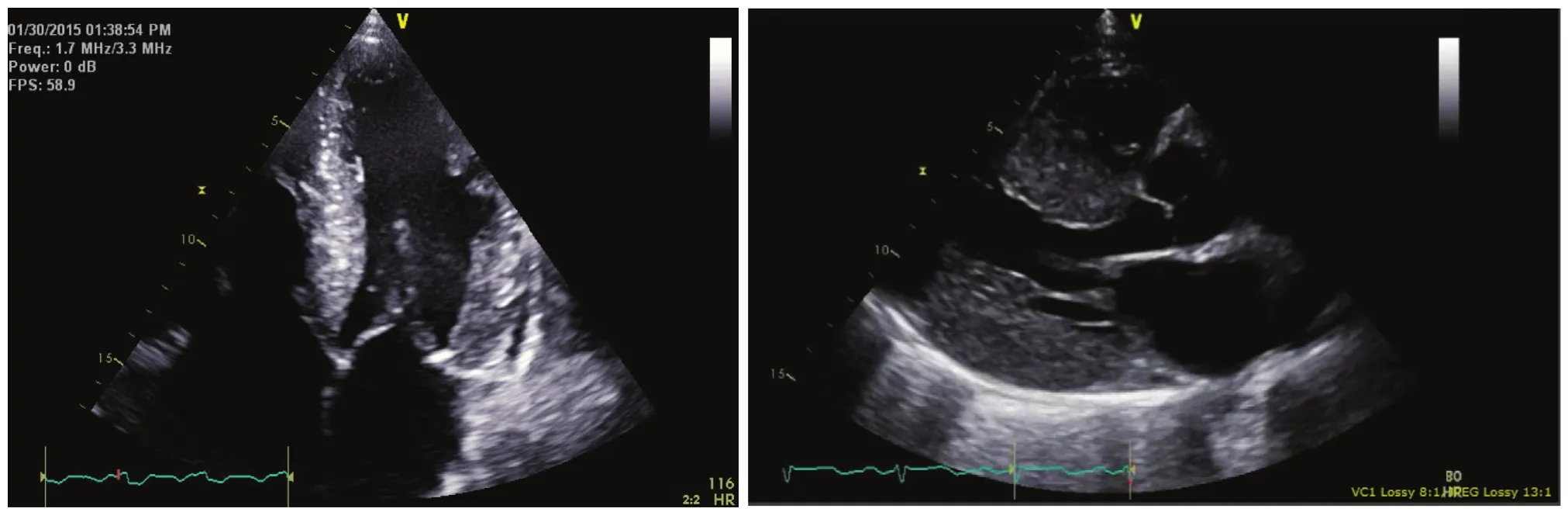
Figure 3 The Left Panel Shows an Echocardiogram in the Apical Four-chamber View in a Patient with Systemic Amyloidosis Related to a Primary Blood Dyscrasia that was Diagnosed by Tissue Biopsy in Multiple Other Organs.
Volume control with diuretics is also fairly diff i cult since patients with this degree of restriction require elevated filling pressures to maintain any reasonable cardiac output. Maintenance of the slow heart rate is important in the treatment of these individuals; however, because conduction abnormalities can result from amyloid protein deposition, caution must be taken to avoid signif i cant bradycardia and heart block.
Digitalis glycosides should be avoided in amyloidosis because of the preferential binding to amyloid protein, and increased clinical effects even at therapeutic blood levels [29].
Conclusion and Take-Home Message
Uncommon cardiomyopathies such as LVNC and cardiac amyloidosis may not be as uncommon as had previously been thought. In fact, these disease states represent a fairly signif i cant portion of individuals seen in an advanced heart failure practice for HFrEF and HFpEF, respectively. It is important to remember these clinical entities when one is evaluating patients with heart failure. A thorough history,including a family history, and physical examination and routine diagnostic tools such as echocardiography continue to be the diagnostic armamentarium of the heart failure cardiologist. Further evaluation of individuals with dilated cardiomyopathy or severe restrictive cardiomyopathy should include CMRI,since this diagnostic tool has proven to be both sensitive and specific for LVNC and very specific for the presence of cardiac amyloidosis.
As genetic testing evolves, and cost of genetic testing falls, the use of routine genetic testing in heart failure and cardiomyopathy may prove to be of substantial benef i t. Furthermore, as our understanding of the microRNA changes in all heart failure patients improves, heart failure patients may be able to have personalized medical therapies to alter their disease progression.
Conflict of interest
The authors declare no conf l ict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial or notfor-profit sectors.
1. Grant RT. An unusual anomaly of the coronary vessels in the malformed heart of a child. Heart 1926;13:273–83.
2. Jefferies JL, Wilkinson JD, Sleeper LA, Colan SD, Lu M, Pahl E, et al.Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: results from the pediatric cardiomyopathy registry. J Card Fail 2015.
3. Samsa LA, Yang B, Liu J. Embryonic cardiac chamber maturation:trabeculation, conduction, and cardiomyocyte proliferation. Am J Med Genet C Semin Med Genet 2013;163C(3):157–68.
4. Jenni R, Oechslin E, Schneider J,Attenhofer Jost C, Kaufmann PA.Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction:a step towards classif i cation as a distinct cardiomyopathy. Heart 2001;86(6):666–71.
5. Xing Y, Ichida F, Matsuoka T,Isobe T, Ikemoto Y, Higaki T, et al.Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab 2006;88(1):71–7.
6. Almeida AG, Pinto FJ. Noncompaction cardiomyopathy. Postgrad Med J 2014;90(1062):208.
7. Botto LD. Left ventricular noncompaction. In: Orphanet Encycolopedia. Paris, France: INSERM;2004. pp. 1–6.
8. Paterick TE, Gerber TC, Pradhan SR, Lindor NM, Tajik AJ. Left ventricular noncompaction cardiomyopathy: what do we know? Rev Cardiovasc Med 2010;11(2):92–9.
9. Towbin JA. Left ventricular noncompaction: a new form of heart failure. Heart Fail Clin 2010;6(4):453–69, viii.
10. Patterson AJ, Zhang L. Hypoxia and fetal heart development. Curr Mol Med 2010;10(7):653–66.
11. Oechslin E, Jenni R. Left ventricular non-compaction revis-ited: a distinct phenotype with genetic heterogeneity? Eur Heart J 2011;32(12):1446–56.
12. Song ZZ. Echocardiography in the diagnosis left ventricular noncompaction. Cardiovasc Ultrasound 2008;6:64.
13. Amir O, Delgado RM, Kar B, Smart FW. The value of cardiac magnetic resonance imaging in the diagnosis of isolated non-compaction of the left ventricle. Isr Med Assoc J 2009;11(5):313–4.
14. Dodd JD, Holmvang G,Hoffmann U, Ferencik M, Abbara S, Brady TJ, et al. Quantif i cation of left ventricular noncompaction and trabe cular delayed hyper enhancement with cardiac MRI: correlation with clinical severity. Am J Roentgenol 2007;189(4):974–80.
15. Sidhu MS, Uthamalingam S, Ahmed W, Engel LC, Vorasettakarnkij Y,Lee AM, et al. Def i ning left ventricular noncompaction using cardiac computed tomography.J Thorac Imaging 2014;29(1):60–6.
16. Stöllberger C, Finsterer J. Left ventricular hypertrabeculation/noncompaction and stroke or embolism. Cardiology 2005;103(2):68–72.
17. Shah KB, Inoue Y, Mehra MR.Amyloidosis and the heart: a comprehensive review. Arch Intern Med 2006;166(17):1805–13.
18. Hassan W, Al-Sergani H, Mourad W, Tabbaa R. Amyloid heart disease: new frontiers and insights in pathophysiology, diagnosis,and management. Tex Heart I J 2005;32(2):178–84.
19. Ruberg FL, Berk JL. Transthyretin(TTR) cardiac amyloidosis. Circulation 2012;126(10):1286–300.
20. Ng B, Connors LH, Davidoff R,Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med 2005;165(12):1425–9.
21. Tanskanen M, Peuralinna T,Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med 2008;40(3):232–9.
22. Falk RH. Diagnosis and management of the cardiac amyloidoses.Circulation 2005;112(13):2047–60.
23. Noël LH, Zingraff J, Bardin T,Atienza C, Kuntz D, Drüeke T. Tissue distribution of dialysis amyloidosis. Clin Nephrol 1987;27(4):175–8.
24. G al R, Korzets A, Schwartz A, Rath-Wolfson L, Gafter U.Systemic distribution of beta 2-microglobulin-derived amyloidosis in patients who undergo long-term hemodialysis. Report of seven cases and review of the literature. Arch Pathol Lab Med 1994;118(7):718–21.
25. A nsari-Lari MA, Ali SZ. Fineneedle aspiration of abdominal fat pad for amyloid detection: a clinically useful test? Diagn Cytopathol 2004;30(3):178–81.
26. M aceira AM, Joshi J, Prasad SK,Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis.Circulation 2005;111(2):186–93.
27. F alk RH, Plehn JF, Deering T,Schick EC Jr, Boinay P, Rubinow A, et al. Sensitivity and specificity of the echocardiographic features of cardiac amyloidosis. Am J Cardiol 1987;59(5):418–22.
28. L indsay S. The heart in primary systemic amyloidosis. Am Heart J 1946;32(4):419–37.
29. R ubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation 1981;63(6):1285–8.
杂志排行

Cardiovascular Innovations and Applications的其它文章
- Strategies to Reduce Heart Failure Hospitalizations and Readmissions:How Low Can We Go?
- Congestive Heart Failure Clinics: How to Make Them Work in a Community-Based Hospital System
- Cardiac Sarcoidosis: Sorting Fact from Fiction in This Rare Cardiomyopathy
- Epidemiological Study of Heart Failure in China
- Noninvasive Hemodynamic Monitoring for Heart Failure: A New Era of Heart Failure Management
- The Evaluation of the Heart Failure Patient by Echocardiography: Time to go beyond the Ejection Fraction