Drug Registration and Approval System in China: Current Status,Controversies and Future Perspectives
2015-05-15ZHAOJunruiNIJingyunSITOUKawengHUYuanjia
ZHAO Jun-rui,NI Jing-yun,SI TOU Ka-weng,HU Yuan-jia*
(State Key Laboratory of Quality Research in Chinese Medicine,Institute of Chinese Medical Sciences,University of Macau,Macao 999078,China)
Drug Registration and Approval System in China: Current Status,Controversies and Future Perspectives
ZHAO Jun-rui,NI Jing-yun,SI TOU Ka-weng,HU Yuan-jia*
(State Key Laboratory of Quality Research in Chinese Medicine,Institute of Chinese Medical Sciences,University of Macau,Macao 999078,China)
Objective To present a comprehensive analysis on the current status and controversies of China’s drug registration and approval system in transition period and to disclose the future direction and trends by analyzing the draft amendment to Drug Registration Regulation (DRR) as well as relevant notices. Methods Literature review,qualitative analysis,and descriptive statistics were applied. Results and Conclusion Current DRR in China seems incompatible and outdated in a number of respects,such as approval timelines,generic drug registration,binding authorization mode,amending clinical protocols,and new drug observation periods. Draft amendments to DRR as well as relevant notices for public comment released by Chinese regulatory authorities show partial responses to current status and controversies. The draft amendment indicates a future direction and tendency for drug registration and approval. On the basis that the China Food and Drug Administration (CFDA) is attempting to improve the efficiency of the approval process,the amendments will further encourage drug R&D and help to restore confidence among both the public and the drug manufacturers.
China; drug registration; drug approval; drug evaluation
1 Introduction
Past several decades have witnessed a series of tremendous changes and improvements in China’s drug registration and approval system. This development course[1]has been described as comprising 4 stages: exploration (1963–1983),development (1984–late 1990s),standardization (2001–2006),and improvement (2007–present).
The day July 10,2007 represents a milestone in the development of China’s pharmaceutical sector. On that day,the State Food and Drug Administration (SFDA),China’s former regulatory administration,issued the Drug Registration Regulation (DRR) which specifies a new regulatory framework for pharmaceutical and biological products[2,3]. On the same day,the former commissioner of the SFDA was executed for accepting bribes from various firms in exchange for drug licenses. Since then,the pharmaceutical regulatory system in China experienced evolvement,SFDA realizes the necessity to update these waves of reform and dramatic development. After years of regulations in that they are becoming incompatible with real situations,such as approval timelines,generic drug registration,binding authorization mode,amending clinical protocols and observation periods.
In this context,the amendment of DRR is given high priority on the agenda of the China Food and Drug Administration (CFDA),current official pharmaceutical authority. On November 13,2013,CFDA published a draft amendment to DRR[4]for public comment. Provincial Food and Drug Administrations (PFDAs) were required to collect comments within a month from pharmaceutical manufacturers and R&D institutes,as well as from other organizations and individuals. On February 19,2014,the Legislative Affairs Office of China’s State Council released a further revised draft amendment to the DRR,with any public comments due by March 23,2014[5]. In addition,the Department of Drug and Cosmetics Registration of CFDA drafted a notice clarifying requirements for applications forpharmaceutical registration; this was released internally for comments on September 11,2014[6].
As the leading representative of “pharmerging”countries,China is forecast to become the world’s second largest pharmaceutical market by 2015[7],and policy changes on drug registration in China therefore attract a great deal of attention worldwide from policymakers,investors,and researchers in the pharmaceutical sector. Although they have not yet taken legal effect,the documents mentioned above disclose most of the likely new rules for Chinese pharmaceutical registration. However,there is no comprehensive analysis of Chinese drug registration system during the transition period,and as the lack of clarity around the possible new rules may as well be confusing for the international community. This article presents an overview of China’s latest drug regulation system,discusses the hottest controversies and comprehensively discloses the new rules that seem likely to govern drug registration in China.
Section 2 of this article introduces China’s latest pharmaceutical regulatory framework. Section 3 presents the facts and figures about Chinese drug registration,and Section 4 discusses the main controversies arising. The main possible changes to drug registration in China are highlighted and comprehensively analyzed in Section 5. Finally,Section 6 presents a concluding summary of the main changes and potential implications for the future.
2 China’s drug regulatory system
China’s regulatory system for the pharmaceutical sector operates at four levels: national,provincial,municipal and county. In 2008,the State Council issued a document canceling direct management in drug regulatory departments below provincial level,revising the vertical supervision system below provincial level to a system of hierarchical management by local governments,with work specified,organized,and supervised by superior administrative departments and health departments at the same level[8].
In March 2013,the State Council announced the establishment of CFDA,which is separate from the Ministry of Health and directly subordinate to the State Council as an authority at ministerial level. The newly established CFDA integrates the relevant responsibilities of the State Council’s Food Safety Office,SFDA,the State General Administration of Quality Supervision,Inspection and Quarantine,and the State Administration for Industry and Commerce[9]. The aim was to remove the overlap in responsibility of various departments,so improving administrative efficiency. Figure 1 outlines the structure of China’s current system of drug regulation.
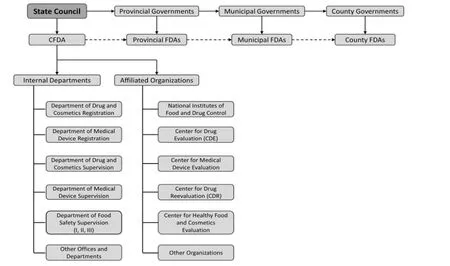
Figure 1 China’s current drug regulatory system
CFDA then began to restructure former SFDA responsibilities,internal departments,and affiliated organizations. In May 2013,a document entitled Provisions of Responsibilities,Departments and Staffing of the CFDA[10]was released,introducing these inner reform plans to the public domain. The changes can be classified under 2 headings.
(1) Internal departments. Relative to the former SFDA’s internal departments,there were 3 main adjustments. First,the former Department of Food Safety Supervision was separated into three parallel Departments (Department I,Department II,and Department III),which were to be responsible for production supervision,circulation supervision,and food safety risk evaluation,respectively. Second,the administration and supervision of drugs and cosmetics were merged,and the Department of Drug and Cosmetics Registration and Department of Drug and Cosmetics Supervision were established. Finally,the former Department of Medical Device Supervision was split into the Department of Medical Device Registration and the Department of Medical Device Supervision.
(2) Functions and duties. Under this restructuring,some responsibilities will be withdrawn or decentralized. To begin,administrative licensing is to be integrated into a single function,to include drug production and Good Manufacturing Practice (GMP) certification,pharmaceutical trade and Good Supply Practice (GSP) certification,and cosmetics production and cosmetics hygiene. Duties related to licensed pharmacists’ continuing education will be transferred to the China Licensed Pharmacist Association. Certain responsibilities will be decentralized to provincial counterparts,including ① GMP certification of drugs and medical devices; ② drug re-registration and licensing of supplementary applications (for drugs) that do not alter the products’ inherent quality; ③ licensing for changes of domestic-made Class III medical devices that do not change inherent quality; ④ licensing of drug contract production; and ⑤ licensing of non-special purpose cosmetics imports.
3 Facts and figures
In order to understand the amendment of the DRR and associated controversies,it is necessary to review facts and figures of Chinese drug registration. Most of the data in this section were retrieved from the Annual Report on Drug Registration and Approval in China[11-13]published by CFDA,and China Drug Review Annual Report[14,15]published by Center for Drug Evaluation (CDE).
It is worth noting that,due to corruption scandals,drug registration in China was extremely disorderly during the years prior to 2007. During this period,over ten thousand application items were approved annually,including more than one thousand new drugs. Since 2007,the SFDA has undergone reorganization,drugs approved to the market have been reexamined,and a more normalized situation has been in place since 2009.
Figure 2 shows the data for registration applications accepted annually and completed by CFDA (2009–2013). In general,the number of accepted applications has increased steadily in the last five years; among the three drug types,chemical drug applications accounted for over eighty percent.
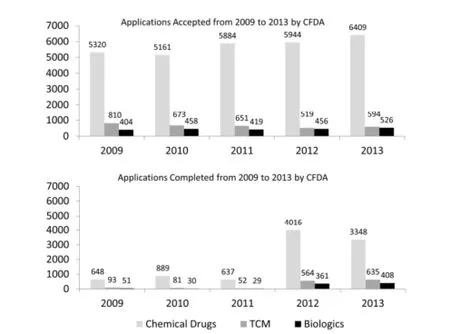
Figure 2 Drug registration applications accepted and completed by China Food and Drug Administration (CFDA) (2009—2013)
However,the number of completed applications is much lower than that of accepted applications,especially in the first three years. Despite a dramatic increase in 2012,the number of finished applications again declined in 2013,which shows that CFDA remains unstable in drug registration and approval work.
Figure 3 discloses the annual completion rates for three types of application over the last five years; the completion rate is calculated as the ratio between number of finished and accepted applications. In the first three years,CFDA figures for drug approval almost stagnated before dramatically accelerating in 2012. The subsequent completion rate showed a slight decline for traditional Chinese medicine (TCM) and biologics,and an obvious decrease for chemical drugs. As a drug type with Chinese characteristics,completion rates for TCM are higher than for the other two drug types in 2012 and 2013.
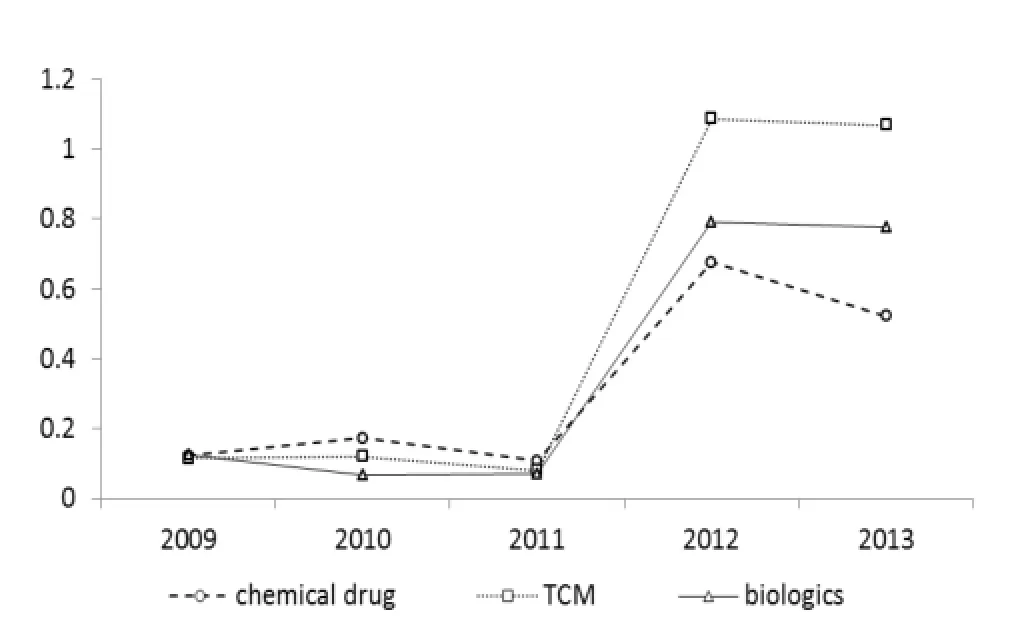
Figure 3 Annual Completion rates of chemical drug,traditional Chinese medicine and biologics applications (2009—2013)
Most complaints from the industry concerning drug registration focus on the long waiting times for technical review. Table 1[15]details average waiting times in months for technical review of chemical drugs. January 2012,December 2012,and December 2013 are the time points at which CDE commenced technical review of certain applications,and reception date refers to the point at which CDE received the technical review request from CFDA. Waiting time is then calculated as the difference between reception date and time of starting technical review. Rows in the table represent different channels of technical review.
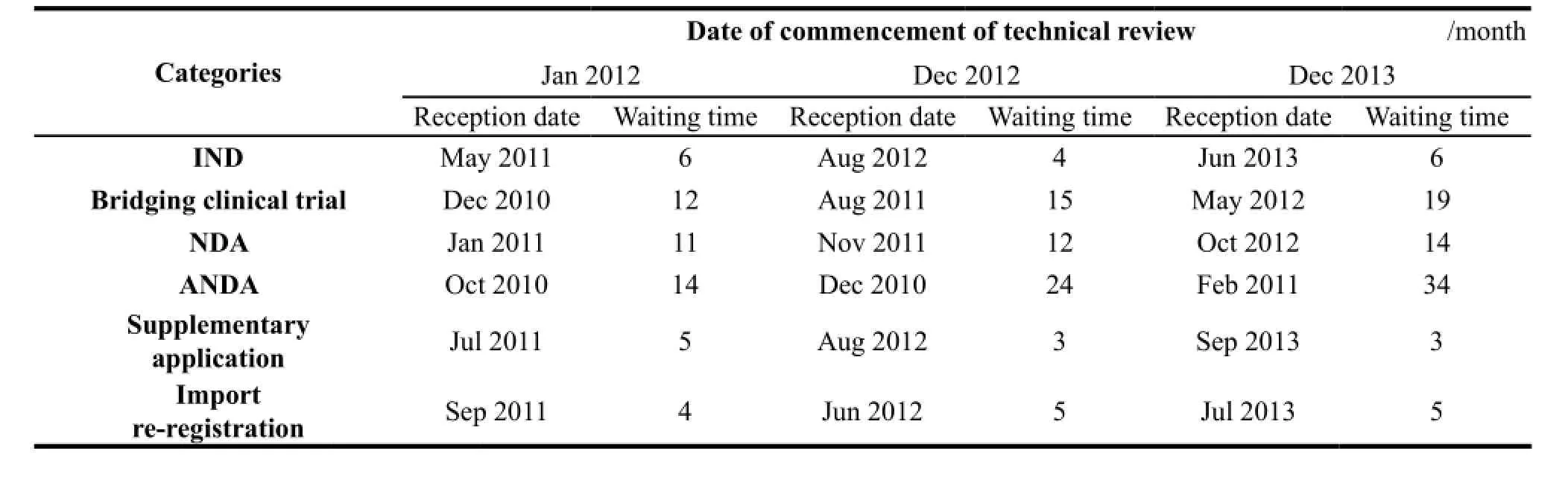
Table 1 Waiting times for technical review of chemical drugs
Waiting times for Investigational New Drugs (INDs),supplementary applications,and import re-registration were relatively steady,but waiting times lengthened in the other three channels,especially for Abbreviated New Drug Applications (ANDAs). It is clear that ANDA waiting times have increased significantly in recent years; these cases may take a couple of years to achieve final approval. Additionally,although CFDA has special review procedures for New Drug Applications (NDAs) to encourage drug innovation,there may still be a wait of one year or more before CDE initiates the technical review.
4 Controversies
During the turbulent transition period,drug registration in China appears to have provoked several controversies,focusing mainly on the following four issues.
4.1 Insufficient review resources
As one of the core affiliated institutions of CFDA,CDE is responsible for conducting technical review of drug in registration and providing technical support for CFDA,which plays a key role in drug registration process. Figure 4 shows the detail structure of CDE.
Due to the special political system of China,the fiscal budget and human resources of CDE are almost entirely allocated and arranged by central government. At present,CDE has only 105 members of staff responsible for technical review[16],whereas the Center for Drug Evaluation and Research (CDER) at the U.S. Food and Drug Administration (USFDA) had a total of 3,603 staff members in 2013[17]. CDE’s resources are apparently insufficient to cope with thousands of registration applications per year,and this lack of resources inevitably undermines the efficiency and quality of technical review of pharmaceuticals in China.
It is difficult to improve this situation within a short time because of the fixed political system,which is a long-term arrangement,and CFDA is not allowed to purchase external evaluation resources directly. In these circumstances,some scholars have suggested that CFDA should dramatically increase application fees for drugregistration in order to be able to access more evaluation resources and ultimately enhance review efficiency and quality,as in the case of the Prescription Drug User Fee Act (PDUFA) in the U.S[18].
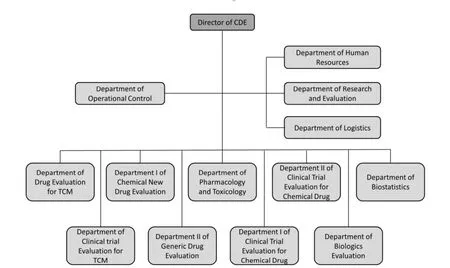
Figure 4 Structure of center for drug evaluation of CFDA
4.2 Over-centralized approval mode
China’s current drug registration and approval system adopts a centralized approach. Specifically,as a ministeriallevel institution,CFDA possesses core approval authority while provincial counterparts are responsible mainly for preliminary checks and dossier submission. Arguably,this can to some extent facilitate CFDA in directly controlling the core stages of the approval process and in responding rapidly to dynamic environments.
However,such an approach has been called into question as over-centralized,especially at a time when drug registration and approval became disordered. For quite a long period,CFDA was unable to strictly abide by the legally required timeline,while applicants could do nothing but wait. And,of course,no one will reimburse applicants for their incalculable losses during the long wait.
In recent years,CFDA has been aware of the significance of “power decentralization”,and really did it in the 2013 restructuring. CFDA selected Guangdong as the pilot province and authorized the Guangdong Food and Drug Administration to conduct technical review and administrative approval in respect of certain registration items[19]. Even so,there are few substantial changes,and CFDA still controls most approval authorities. In short,there is still a long way to go if the over-centralized approval mode is to change.
4.3 Disordered administration in generic drug registration
In 2013,CFDA accepted 2,427 generic drug applications,accounting for more than a third of chemical drug applications. Across all generic drug applications,61.6% attempted to duplicate marketed drugs that had already been imitated more than ten times[15]. While this phenomenon can largely be attributed to current developmental level of China’s pharmaceutical industry,characterized by a lack of innovation and the prevalence of low-level repeated imitation,CFDA can be criticized on a number of counts with regard to generic drug administration.
First,some generic drug registration procedures are inappropriate and significantly hinder registration process. For example,to win CFDA approval,a bioequivalence study of generic drug registration is first required. Some scholars doubt the necessity of this approval process andsuggest a filing system similar to that in the U.S.,where a bioequivalence study of generics is required only for filing in the official recording system without any need for administrative approval[20]. On the other hand,some researchers have argued that it is unreasonable to require onsite production inspection prior to a bioequivalence study,as formulae and the manufacturing processes may be adjusted on the basis of the results of that study[20].
Second,there is a low threshold in China for generic drug registration[21]. At present,marketing approval for a generic drug incurs no charge for the applicants. Consequently,the cost for manufacturers of submitting a registration application is low,and irrational applications arise. In addition,CFDA has no pre-screening procedures to identify the clinical value of the drug in question. Under pre-screening procedures,low value applications could be suspended or rejected while the pace to market of high value applications could be accelerated by establishing special review procedures.
Third,CFDA has seldom offered guidance to generic manufacturers,making it difficult for the industry to follow and execute the policies of CFDA effectively. Recently,CFDA has recognized the significance of policy transparency and enhanced communication with the industry,releasing two lists of excessively imitated drug categories on its official website on September 16,2014 and November 14,2014,respectively[22,23]. Reducing the industry’s blind and excessive imitation on certain drugs can help to reduce the pressure of technical review for CFDA.
4.4 Outdated “binding authorization mode”
In China,drug marketing authorization is only licensed to the pharmaceutical manufacturers with production authorization; this is the so-called “binding authorization mode” adopted by CFDA. This means that any drug manufacturer,as the marketing subject,can easily be supervised by CFDA and has relatively wide scope for risk-taking.
However,negative influences have accompanied the development of the pharmaceutical industry. For one thing,R&D institutions have no authority to market the drug they developed. And because they cannot therefore get a reasonable return,this will almost certainly impair the motivation to innovate and hinder the transfer of technology outcomes. Additionally,for manufacturers,a new production line must be established and certified in order to launch a drug to the market,even where the drug has been massively imitated or other manufacturers have suspended production of the same drug. The inevitable consequence is excessive but idle production capacity and low-level repeated production across the whole industry.
In recent years,scholars and industry experts in China have advocated the enforcement of a “separated authorization mode” to tackle these problems,and a marketing authorization holder (MAH) system was seen as the solution. SHAO Rong[24]used a demand-supply model of institutional change to analyze the necessity and feasibility of such a system and concluded that a MAH system should be established in China. On the other hand,CHEN Yong-fa[25]has pointed out the risks in a MAH system,urging particular caution in respect of contract production,post-marketing safety,and consumer protection processes.
5 Amendments
In response to the growing controversy and debate over drug registration,the CFDA amended and released new guidelines/regulations for this issue based on relevant draft amendments[4,5],official drafting explanation[4]and official notice[6]. These changes focused mainly on the following aspects:
5.1 Elimination of discrepancies between DRR and Chinese patent law
As stated in the 2007 version of DRR,there are currently two provisions regarding drug patent protection in the registration process. Article 18 stipulates that when an applicant submits a drug registration request,the formulae,manufacturing processes and/or uses of that drug,patent information and its ownership status,as well as the non-infringement statements,shall all be provided to the CFDA. If patent disputes arise while the application of drug registration is proceeding,they must be settled under relevant patent laws and regulations. As for generic drug manufacturers,in accordance with Article 19,they can only submit the registration requests no more than two years before the expiration of corresponding drug patents. Generic drugs which are approved by the CFDA will be given a valid Drug Approval Number along with some other certificates right after the expiry date of the patent.
In the latest draft amendments,some adjustments have been made on both the provisions and authorization procedures. First,it is suggested that the phrase “during the registration process” in Article 18 and the two-year limit for Article 19 shall be removed. Secondly,approved genericdrugs will receive Drug Approval Number and relevant certificates in advance,but these documents will only come into effect after the drug patent expires.
According to the Drafting Explanation[4],such modifications aim to eliminate contradictions and discrepancies between DRR and China’s Patent Law. In 2008,patent infringement exemption for drugs has been extended such that patents of drugs,medical apparatus and instruments can be used for the purpose of providing necessary information for administrative approval[26]. This new exemption is similar to the “Bolar Exemption”,a provision which is widely used to encourage drug R&D. Under these circumstances,patent disputes during the registration process describe in Article 18 no longer exist and the phrase can be omitted.
Another issue that raises much concern is the time limit on generic drug applications. Since the evaluation and approval time take much longer than before,the two-year period is not applicable to current applications anymore. As a consequence of these delays,the rights of generic manufacturers to market the drug on time are deprived,implying that patented drugs gain an extended patent life automatically.
5.2 Addition of supplemental application
Under current DRR,clinical trial applications submitted to the CDFA cannot undergo any changes. If there are alterations to certain items such as formulae,manufacturing processes or production sites,the applicant can only choose to withdraw the application and file an entirely new one.
In order to meet the needs of the pharmaceutical companies,the draft amendments added Article 50 to DRR,enabling the undermentioned changes to be made during clinical trials in the form of supplemental application:
(1) Applicant changes;
(2) Modifications on manufacturing process,formulae,strength in chemical drugs or biologics prior to Phase III clinical trials;
(3) Relocation of manufacturing sites prior to Phase III clinical trials;
(4) Changes to the preparation processes and strength of TCM.
Alterations to the first three items require the presentation of relevant certificates and research data to the CFDA while changes to the last item have to be conducted pursuant to Complementary Provisions for TCM Registration[28].
The Drafting Explanation[4]pointed out that addition of the new article further encourages drug R&D and innovation by providing pharmaceutical companies with a greater level of flexibility to react to the rapidly changing environment.
5.3 Adjustments of “observation period”
When a new drug receives approval for production,it is subjected to an observation period of no more than five years to ensure its safety. During this period,CFDA shall not accept new registration request from any manufacturers to produce,import,or to change the dosage forms of that particular drug. In addition,applications which have been accepted but not yet approved will also be returned. Nevertheless,this restriction do no applies to clinical trial applications of the same drug which have been approved before observation period begins. They will still be allowed to proceed to the subsequent steps in drug registration processes,and are able to obtain marketing authorization as long as they meet the requirements listed in the DRR.
In the draft amendments,CFDA proposed that when a new drug enters its observation period,already accepted but not-yet-approved applications from other manufacturers can continue with their registration processes. A similar rule applies to imported drug which gains its first marketing authorization,and there is even one more option for applicants who have submitted their applications; they can either choose to carry on with the registration procedures or to withdraw and replace their accepted application with a generic one.
As mentioned in the Drafting Explanation[4],the purpose of these adjustments is to solve the controversy on“registration application returning” and to further stimulate drug R&D. The current ordinance has created a hostile atmosphere in the industry because all drug manufacturers have to compete with each other and strive to gain clinical trials approval ahead of the first NDA issuance. Under the new provision,there are more opportunities for applicants and it helps to build a healthy competitive environment for pharmaceutical innovation.
5.4 Modifications on generic drug application process
Before starting a bioequivalence study,a generic drug must go through and pass several stages of inspection. Once a generic drug application is submitted,PFDA will carry out an on-site inspection on drug R&D conditions and itsproduction site. In addition,the applicants have to bring forth the raw data together with quality specifications of the drug. In the meantime,three batches of products will be collected as samples and sent to the drug testing institutes for further examination.
In fact,the practice of conducting production site inspection at an early stage of generic drug application procedures causes some problems,such as resource wastage and low quality checks,and is also detached from technical review and GMP inspection. Stated in the Drafting Explanation[4],the new draft amendment intends to reduce conflicts and optimize generic drug application procedures,therefore it has postponed both the inspections on manufacturing process and quality specifications such that they will be conducted on completion of the bioequivalence study. Putting off the inspections to a later stage enables manufacturing processes to be optimized in line with the results of bioequivalence study.
5.5 Miscellaneous revisions of DRR
Aside from the major changes that we have mentioned,other subtle amendments include:
(1) All non-clinical safety evaluations must be conducted at GLP-certified institutes.
(2) The starting time for technical review is clearly defined as the day on which CDE commences a technical review.
(3) The draft amendment added two circumstances in which applications would be rejected by CFDA: (a) where endangered animals and plants are used as raw materials and sustainable obtainment cannot be ensured; and (b) where production site inspections are not applied within six months.
(4) SFDA was renamed as CFDA,and the National Institute for the Control of Pharmaceutical and Biological Product (NICPBP) was renamed as the National Institute for Food and Drug Control (NIFDC).
According to the Drafting Explanation[4],the first two measures are designed to enhance research quality on non-clinical safety evaluations and to address the ambiguity regarding technical review starting time respectively. Introduction of item 3a aims to avoid excessive exploitation of endangered animals and plants by pharmaceutical companies,whereas item 3b is employed to improve the management mechanism for production site inspections.
5.6 Rearrangement of internal operating procedures
The CFDA Department of Drug and Cosmetics Registration internally released a notice[6]for comment on September 11,2014,regarding the criteria for accepting pharmaceutical registration application. The notice focused on internal operating procedures of drug registration and application acceptance,particularly chemical drugs,as well as some procedural changes. Significant modifications are listed as follow:
5.6.1 New drug application
In NDA,marketing approval can only be applied after the completion of clinical trials. But now 4 drug categories can be exempted from clinical trial applications,implying that applicants can directly apply for marketing approval. The 4 categories are normal or specific immune globulins for intramuscular injection,human albumins,multiple electrolytes injections and blood volume expanders.
Moreover,CFDA outlined new criteria for the registration of new indications for an approved drug: only the applicants who hold the Drug Approval Number,Import Drug License or Pharmaceutical Product License of that drug are entitled to submit the new indication application.
5.6.2 Generic drug application
The application procedures for chemical drug category VI have slightly been simplified. If the first generic of a pioneer drug meets all the requirements after evaluation,it can be marketed immediately without going through clinical trials.
5.6.3 Import drug application
Based on current registration rules,when a foreign manufacturer submits clinical trial application for a new drug which has never been marketed (drug category I),Certificate of Pharmaceutical Product (CPP) issued by the exporting country should be handed in to CFDA simultaneously. This unreasonable measure led to complaints and oppositions from foreign manufacturers and therefore the CFDA suggested extending the submission deadline for this document to the day on which application for marketing approval is made.
5.6.4 Others
Apart from emphasizing associated applications between pharmaceutical preparations and their active pharmaceutical ingredient (API),CFDA has given clearguidelines on operational procedures as well as the requirements for clinical trial exemptions. The practical processes related to supplementary applications are also explicitly presented,such as changes in manufacturing address,reduction of claimed efficacies or therapeutic indications,and cancellation of trade names.
6 Conclusions
In the recent past,China’s current pharmaceutical regulatory system has undergone a series of changes and transformations. To resolve problems associated with drug registration and approval processes,CFDA must address a number of controversies,deal swiftly with feedback,and conceive a new scheme for handling drug regulation.
In the near future,a formally amended DRR needs to adapt to the current level of development of the imitationoriented Chinese pharmaceutical industry while remaining responsible for encouraging drug R&D and innovation. It is a positive feature of the draft amendment to DRR that it notes the efforts of CFDA to solve long-standing disputed issues such as the overlong review process,over-centralized approval mode,the reversed procedure for production onsite inspection,and a weak patent linkage system. However,some critical issues still seem incapable of resolution within a short time,including insufficiency of review resources,the binding authorization mode,and double approval of generics.
From our perspective,several changes may need to be further made on current system. Firstly,in order to fulfill the acceleration of registration process,CFDA can purchase external review resources to reduce the burden of CDE,for example,outsourcing low-risk work to competent provincial FDAs,universities or research institutes. Secondly,CFDA may consider improving the threshold of generic drug application,at the same time,establishing fast track mechanism for new important generics. This will contribute to optimizing the imitation-oriented pharmaceutical industry structure in China. Thirdly,certain registration procedures should be further simplified,such as approval of bioequivalence study,the clinical trial of import drugs that has already been marketed in other countries. Fourthly,the feasibility of some new mechanisms should be explored and considered,for example,the implementation of MAH system,as well as the increase in drug registration and review fee.
While this round of amendments to the current drug registration and approval system has not yet been finalized,the draft amendment at least indicates a future direction and tendency for drug registration and approval. On the basis that CFDA is attempting to improve the efficiency of the approval process,the amendments will further encourage drug R&D and help to restore confidence among both the public and the drug manufacturer.
[1]ZHANG Xiao-fa. Development course and analysis of China’s drug registration and approval system [J]. China Journal of Chinese Materia Medica,2009,34 (20): 2685-2687.
[2]ZHAO Nai-qing,YAO Chen,CHEN Jie. On the amendments of China’s provisions for drug registration [J]. Drug Information Journal,2008,42 (5): 467-475.
[3]LIN Bing-bing. A snapshot of the evolution of pharmaceutical regulations in China and Hong Kong [J]. Drug Information Journal,2010,44 (5): 633-639.
[4]China Food and Drug Administration. Notice on seeking opinions for draft amendment to Drug Registration Regulation [EB/OL]. http://www.sda.gov.cn/WS01/CL0778/94158.html,2013-11-12.
[5]Legislative Affairs Office of the State Council. Notice on further seeking opinions for draft amendment to Drug Registration Regulation [EB/OL]. http://www.chinalaw.gov. cn/article/cazjgg/201402/20140200394953.shtml,2014-02-14.
[6]Securities Times. CFDA plans to improve the work of drug registration [EB/OL]. http://www.stcn.com/2014/0919/1172 8943. shtml,2014-09-19.
[7]In-PharmaTechnologist.com. China to claim second place in drug market by 2015,says IMS Health [EB/OL]. http://www. in-pharmatechnologist.com/Regulatory-Safety/China-toclaim-second-place-in-drug-market-by-2015-says-IMS-Health,2010-11-17.
[8]Pharmaceutical Law Institute of Tsinghua University. Reports on reform of China’s Drug Administration Law: Building a Twenty-first Century Pharmaceutical Regulatory System. Available at: https://www.pharmamedtechbi.com /~/media/Supporting%20Documents/Pharmasia%20News/2013/July/Tsing hua_Regulatory_report_062013.pdf,2013-06-19.
[9]Xinhua News Agency. China to elevate food,drug agency to general administration [EB/OL]. http://news.xinhuanet.com/english/china/2013-03/10/c_132221729.htm,2013-03-10.
[10]The Central Government of People’s Republic of China. Provisions of responsibilities,departments and staffing of China Food and Drug Administration [EB/OL]. http://www.gov.cn/zwgk/2013-05/15/content_2403661.htm,2013-05-15.
[11]China Food and Drug Administration. 2009 annual report on drug registration and approval in China [EB/OL]. http://www.sda.gov. cn/WS01/CL0027/54135.html,2010-09-26.
[12]China Food and Drug Administration. 2010 annual report on drugregistration and approval in China [EB/OL]. http://www.sda.gov. cn/WS01/CL0236/65856.html,2011-10-09.
[13]China Food and Drug Administration. 2011 annual report on drug registration and approval in China [EB/OL]. http://www.sda. gov. cn/WS01/CL0236/75250.html,2012-9-29.
[14]Center for Drug Evaluation. 2012 China drug review annual report [EB/OL]. http://www.cde.org.cn/news.do? method=viewIn foCommon&id=313135,2013-06-04.
[15]Center for Drug Evaluation. 2013 China drug review annual report [EB/OL]. http://www.cde.org.cn/news.do? method=viewIn foCommon&id=313280,2014-03-06.
[16]Center for Drug Evaluation. List of CDE review staff (November 2014) [EB/OL]. http://www.cde.org.cn/personal. jsp,2014-11-01.
[17]U.S. Food and Drug Administration. Distribution of full-time equivalent (FTE) employment program level [EB/OL]. http://www.fda.gov/downloads/aboutfda/reportsmanualsforms/reports/budgetreports/ucm301553.pdf,2013-07-06.
[18]TANG Jian-yuan,ZHAO Zhi-heng. Dcussion on China drug administrative charges with reference to U.S. PDUFA [J]. Chinese Pharmaceutical Affairs,2013,27 (6): 564-567.
[19]China Food and Drug Administration. CFDA authorizes Guangdong as pilot province in respect of drug evaluation and approval [EB/OL]. http://www.sda.gov.cn/WS01/CL0 194/76115. html,2012-11-15.
[20]CHEN Zhen,ZHANG Pei-pei. Discussion on strategy of chemistry,manufacturing and controls evaluation for chemistry drug [J]. The Chinese Journal of Clinical Pharmacology,2011,27 (10): 812-816.
[21]DING Jin-xi,LIU Yang-yang,HU Xue-ying,et al. Comparative analysis on the marketing approval system of the generic drugs between China and the USA [J]. Chinese Journal of Pharmaceuticals,2014,45 (2): 190-198.
[22]China Food and Drug Administration. First list of excessively imitated drug categories [EB/OL]. http://www. sda.gov.cn/WS01/CL0087/106390.html,2014-09-12.
[23]China Food and Drug Administration. Second list of excessively imitated drug categories [EB/OL]. http://www.sda.gov.cn/WS01/CL0087/109241.html,2014-11-14.
[24]SHAO Rong,ZHENG Lan,HU Chen-xi,et al. Necessity and feasibility analysis of establishing marketing authorization holder system in China: based on an institutional change demand-supply model [J]. China Pharmacy,2014,25 (33): 3076-3080.
[25]CHEN Yong-fa,ZHAO Min,ZHAO Yan-jiao,et al. Theoretical analysis on system risks of marketing authorization holder system in China [J]. Chinese Journal of New Drugs,2013,22 (24): 2847-2851.
[26]China Food and Drug Administration. Provisions for Drug Registration (SFDA Order No.28) [EB/OL]. http://eng. sfda.gov. cn/WS03/CL0768/61645.html,2007-07-10.
[27]State Intellectual Property Office of China. Patent Law of People’s Republic of China [EB/OL]. http://english.sipo. gov. cn/laws/lawsregulations/201101/t20110119_566244.html,2008-12-27.
[28]China Food and Drug Administration. Complementary Provisions for TCM Registration [EB/OL]. http://www. sda.gov.cn/WS01/CL0055/27432.html,2008-01-07.
* Corresponding author: HU Yuan-jia,assistant professor. Major research area: medicinal administration. Tel: (853) 88228507,E-mail: yuanjiahu@umac.mo
杂志排行

亚洲社会药学杂志的其它文章
- Toxic TCM Supervision: Current Situations and Countermeasures
- WHO Prequalification of Medicines Program: Technical Assistance Effect
- Analysis of TCM Patented Technology Based on the IPC Classification System
- Detection and Analysis on Outlier of the Average Medical Expense in China
- A Study on the Contract Research Organization
- An Empirical Study on Model of Consumers’ Initial Trust in Online Pharmacies