拓扑异构酶Ⅰ和Ⅱ双重抑制剂的研究进展
2015-05-15盛春泉董国强第二军医大学药学院药物化学教研室上海200433
蒋 琰,盛春泉,董国强(第二军医大学药学院药物化学教研室,上海 200433)
・综述・
拓扑异构酶Ⅰ和Ⅱ双重抑制剂的研究进展
蒋 琰,盛春泉,董国强(第二军医大学药学院药物化学教研室,上海 200433)
拓扑异构酶(topoisomerases,Tops)是参与调节细胞内DNA复制、转录、重组和修复等过程的必需酶。Tops分为TopⅠ和TopⅡ,两者通过DNA切断和连接,维持DNA正常拓扑结构和代谢过程。由于Tops在DNA代谢过程的重要作用,干扰Tops的催化活性或者诱导产生Tops介导的DNA损伤已经成为抗肿瘤治疗的重要策略。Tops已经成为最重要的抗肿瘤靶点之一。综述近年来Tops双重抑制剂的研究进展。
TopⅠ;TopⅡ;双重抑制剂;抗肿瘤药物
拓扑异构酶(topoisomerases,Tops)抑制剂是抗肿瘤药物当中的一个独特分支,拥有着广阔的市场和较好的临床治疗效果。这些药物主要分为两类:TopⅠ抑制剂和TopⅡ抑制剂。
以Tops为靶点的药物大多发展于20世纪六七十年代,早期发现的此类药物以TopⅡ为靶点,代表药物为多柔比星、米托蒽醌、依托铂苷、玫瑰树碱等。后来,美国化学家Wall等[1]从我国珙桐科(Nyssaceae)植物喜树(Cam totheca acum inate)中提取得到喜树碱,霍普金斯大学Hisang教授[2]发现其能够特异性抑制TopⅠ,从而有效抑制肿瘤生长。之后对喜树碱进行深入的结构改造,发现了一系列替康类药物,其中以拓扑替康、伊立替康、SN-38为代表,它们对血液型肿瘤有较好的疗效。
然而,单一靶点的Tops抑制剂有着骨髓抑制、胃肠道毒性和可能导致白血病等副作用,且该类药物因一种酶的抑制导致了另一种酶的过量表达,易产生耐药性。TopⅠ/TopⅡ双重抑制剂可同时作用在细胞周期的两个关键酶,有助于提高药物抗肿瘤活性,降低耐药性,为开发新型抗肿瘤药物提供了新思路[3]。本文综述近年来作用较为突出的、拥有TopⅠ/TopⅡ双重抑制机制的化合物,其中一类是已多次报道并有更深入研究的药物,另一类是最新报道的天然产物及其合成的结构衍生物。
1 TopsⅠ和Ⅱ双重抑制剂
1.1 tafluposide(F11782) tafluposide(F11782,2)是依托铂苷(etoposide,1)的衍生物(图1),是目前发现的TopⅠ/TopⅡ双重抑制剂中机制最为清楚的化合物,其在糖苷键上连有的2个五氟苯酚基,可以缓和化合物与TopⅡ的作用强度。2004年,K ruczynski等[4]确定tafluposide的作用靶点为TopⅠ和TopⅡ,同时发现当该化合物与顺铂联合用药后,有助于长久保持药物敏感性,因此K ruczynski等认为当tafluposide与其他类抗肿瘤药物联合使用可以保持药物敏感性,降低肿瘤耐药性的发生率。后来,Bailly等[5]证实了该化合物对人白血病细胞株HL-60有激活线粒体凋亡的级联作用。

图1 tafluposide和etoposide结构
1.2 batracylin(NSC320846) batracylin(3)是玫瑰树碱(4)的类似物(图2),由Leverkusen合成,后来被选为美国国立癌症研究所(NCI)发展肿瘤治疗计划的测试药,在后来的研究中发现了其TopⅡ抑制活性[6,7]。2007年,Rao和Pomm ier[8]在TopⅠ和TopⅡ介导的Top-DNA裂解复合物检测实验中,发现batracylin在TopⅠ缺陷细胞和依托泊苷抗性细胞中含量只是部分减少,从而确定该化合物是TopⅠ/TopⅡ的双重抑制剂。此外,研究中还发现该化合物比依托铂苷和喜树碱拥有更长的半衰期,使得其比传统的Tops抑制剂切割复合物拥有更为缓慢的可逆性,降低了耐药性和毒副作用。
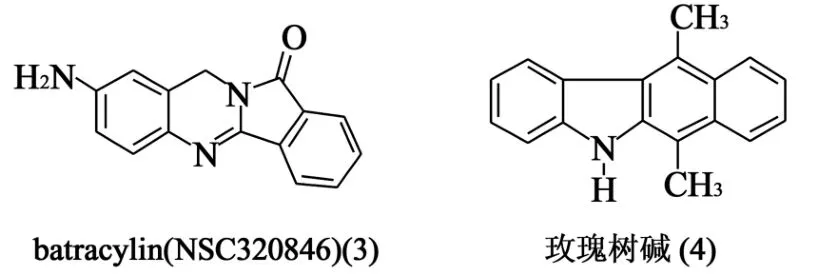
图2 batracylin和玫瑰树碱结构
1.3 吩嗪类衍生物XR11576和XR5944 21世纪初期,拥有抗肿瘤活性的吩嗪类药物陆续被发现,其中XR11576(M LN576,5)和XR5944(M LN944,6)药效最为显著(图3)。
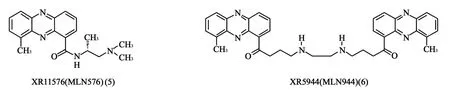
图3 吩嗪类衍生物结构
Lew is和他的同事[9]在针对小鼠多肿瘤细胞抑制实验中发现,XR11576比喜树碱和依托泊苷的体外抗肿瘤活性更强。而且,XR11576具有较好的口服生物利用度,Ⅰ期临床试验的药动学实验证实,当每3周的口服剂量为120mg/d时,该药发挥药效和控制毒性最为理想[10]。
XR5944在临床试验上的研究结果表明,它比目前常用的Tops抑制剂,如多柔比星、拓扑替康、喜树碱等活性更强,Ⅱ期临床试验因为缺少毒性和药动学的关系研究没有确定其使用剂量[11],还需要对药物产生的体内和体外变异进行更多的研究。
1.4 灵菌红素 灵菌红素(prodigioson,7)是一种由微生物S.marcescens和Streptomyces产生的天然红色素,有促进细胞凋亡、对抗肿瘤细胞的作用。Montanor等[12]发现该化合物与DNA嵌入药物氯喹类似,与DNA双链之间的碱基对竞争结合,松弛DNA双螺旋结构。当灵菌红素与DNA结合时,这种结合在AT和GC区是没有区别的,这与很多干扰Tops催化活性的抗肿瘤药物类似。他们同时也验证了该化合物是TopⅠ和TopⅡ双重抑制剂。灵菌红素在体外细胞毒性实验中分别对Jurkat、SW-620和拉莫斯肿瘤细胞株的IC50值分别为225、275和400 nmol/L[13,14]。当与灵菌红素的其他两种衍生物在相同实验条件下对比后,Montanor等认为该化合物C-6甲氧基和吡咯母环为化合物发挥抗肿瘤活性的必需结构(8,9),见图4。
1.5 姜黄素 研究发现姜黄素(图5、10)可以影响转录因子和酶的生理活性,这其中就包括了TopⅠ和TopⅡ[15]。López-Lázaro和他的同事[16]通过在K562白血病细胞中研究姜黄素对TopⅠ和TopⅡ的影响时发现,无论姜黄素浓度为多少,喜树碱诱导的TopⅠ-DNA裂解复合物总量不变。在TopⅡ抑制剂依托泊苷上也有类似的现象出现,可见姜黄素并没有影响到TopⅠ和TopⅡ对DNA的催化活性。同样在K562细胞中,喜树碱与姜黄素联合用药的药效虽强于各自独立用药,却小于两种化合物分别使用的药效总和,这与依托泊苷就有所不同。依托泊苷与姜黄素对各自产生了增强作用[17]。这与经典Tops抑制剂的作用不尽相同。作为有前景的肿瘤化学预防药物,针对姜黄素吸收较差且代谢较快等[18]特点进行的脂质体等剂型改造成为近几年的热点。
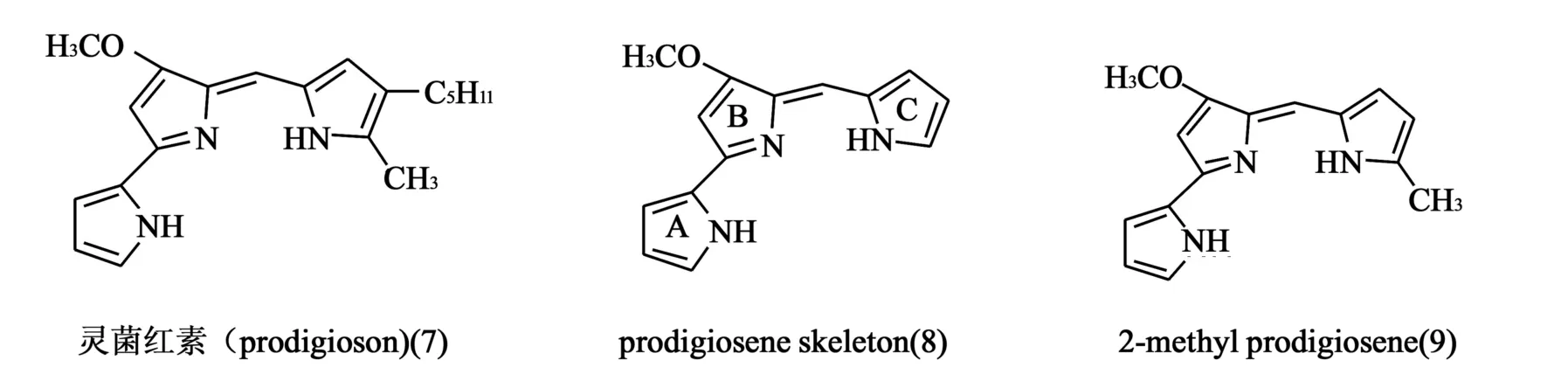
图4 灵菌红素及其衍生物结构
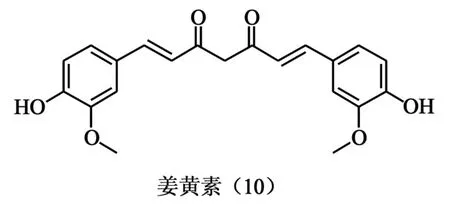
图5 姜黄素结构
1.6 紫椴树皮提取的有效成分 紫椴树是传统的药用植物,其有效成分有镇静、抗焦虑、利尿等作用。2008年,Choi等[19]从紫椴树皮中提取出6个天然化合物(11~16),研究化合物对拓扑异构酶的DNA解螺旋活性的抑制。结果显示,化合物12和13对两种TopⅠ和TopⅡ均有较强的抑制活性,结构见图6。在20μmol/L的浓度下,两种化合物对TopⅡ的DNA解螺旋强于对照药依托泊苷,在100μmol/L的浓度下,对TopⅠ的DNA解螺旋强于对照药喜树碱,抑制率分别为74%和92%。
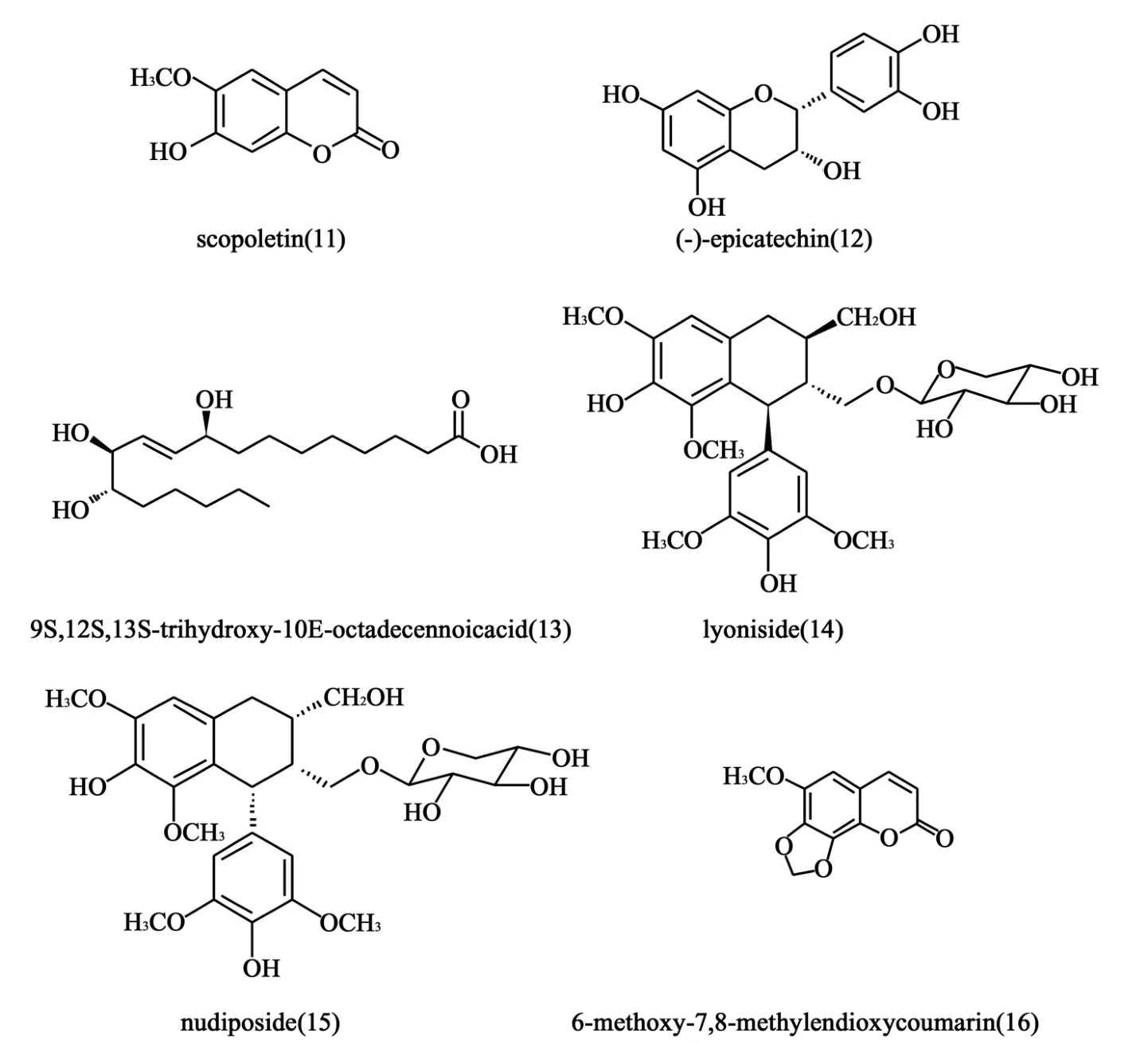
图6 紫椴树皮提取的有效成分结构
1.7 Topopyrones类似物 Topopyrones为真菌Phoma sp.BAUA2861和Penicillum sp.BAUA4206中提取发现的一系列平面蒽醌类化合物[20,21]。这些化合物在结构上与多柔比星、米托蒽醌类似(图7),作用机制上也有相似之处,均为DNA嵌入剂。早期发现认为这些化合物仅为TopⅠ抑制剂。在2008和2010年,Khan和Hecht等[22]发现注入该结构类似物的大多数人白血病细胞停滞在G1期,并不能进入G2/M期,因此他们猜测topopyrones的靶标不只是TopⅠ,后经进一步研究发现,在5μmol/L和20μmol/L浓度时,该类化合物稳定TopⅡ-DNA裂解复合物的效果同依托泊苷相当,从而证明了TopⅠ和TopⅡ均为该药物靶标[23]。

图7 topopyrones类似物结构
1.8 黄酮类及黄酮醇类衍生物 2010年,López-Lázaro等[24]对黄酮类化合物quercetin(21)、apigenin(22)、fisetin(23)和myricetin(24)(图8)在人K562白血病细胞中进行细胞TARDIS鉴定,同时以依托泊苷和喜树碱作为阳性对照药物,发现这些化合物在不同浓度和曝光时间下,21、22和23并没有诱导出TopⅠ-DNA复合物,而且TopⅡ-DNA复合物表达量也不高,只有20在曝光6~24 h的条件下,能够高表达Top-DNA复合物。有趣的是,在XTT基础上的色度比较表明,23虽没有像24一样嵌入DNA分子,但却对两种Tops均有催化抑制活性。通过与其他针对Tops的药物比较,他们认为这4种黄酮类衍生物5位的羟基是发挥TopⅠ和TopⅡ毒性抑制作用的必需基团,而5位没有羟基的结构则表现出拓扑异构酶的催化抑制作用。
1.9 BN 80927(25) 以喜树碱为先导化合物,通过对E环结构改造得到活性更强、代谢稳定性更好的同源类似物高喜树碱(homocamptothecin,hCPT)。2004年,Demarquay等首次报道了新的hCPT——BN80927[25](图9)。Lavergne和Demarquay经DPC和ICT实验发现与hCPT类化合物SN-38相同,且可以稳定TopⅠ-DNA裂解复合物[26]。进一步研究表明,化合物25也可以作用于TopⅡ,抑制TopⅡ活性,但不能稳定TopⅡ-DNA裂解复合物。通过在TopⅠ抗性细胞KBSTP2中加入化合物25的研究发现,该化合物可能属于TopⅡ催化性抑制剂而非毒剂[27]。后来又通过体外实验,验证了本药物活性强于SN-38和依托铂苷(VP-16)。此外,它也可以同时针对G0~G1期的肿瘤细胞,这是喜树碱和其他hCPT类似物所没有的。因此,BN80927被认为是将来可被研究和实用于临床的有潜力的新型药物。
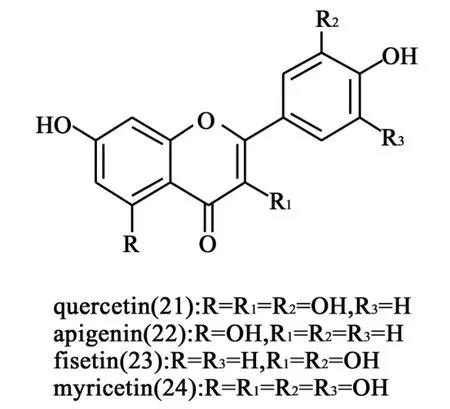
图8 黄酮类及黄酮醇类衍生物结构

图9 BN80927结构
1.10 2,4,6三取代吡啶类似物 2007年,Basnet等[28]合成了一系列的2,4,6三取代吡啶类化合物(图10)来研究该结构的构效关系。化合物26~42被筛选出来进行进一步的拓扑异构酶双重抑制活性的研究,并对其中的35~37进行了细胞毒性评估。在TopⅠ抑制实验中(CPT为阳性对照),35~37和39对TopⅠ具有显著抑制作用,但对TopⅡ抑制作用不强。对TopⅡ抑制实验(依托泊苷为阳性对照)的结果显示,38、40~42的实验结果刚好与前面的结论相反。针对37~42的毒性评估测试(依托泊苷为阳性对照),得到它们针对人4种肿瘤细胞(A549、SK-OV-13、SK-MEL-2、HCT15)的检测结果(表1)。结果表明,2,4′-和2,2′-的联吡啶部分和b,c,d,e,g,h取代基连接对于活性是必需的,而2,4′-和2,3′-的联吡啶和b,c,d取代基连接时,则更多地表现了较强的细胞毒作用。
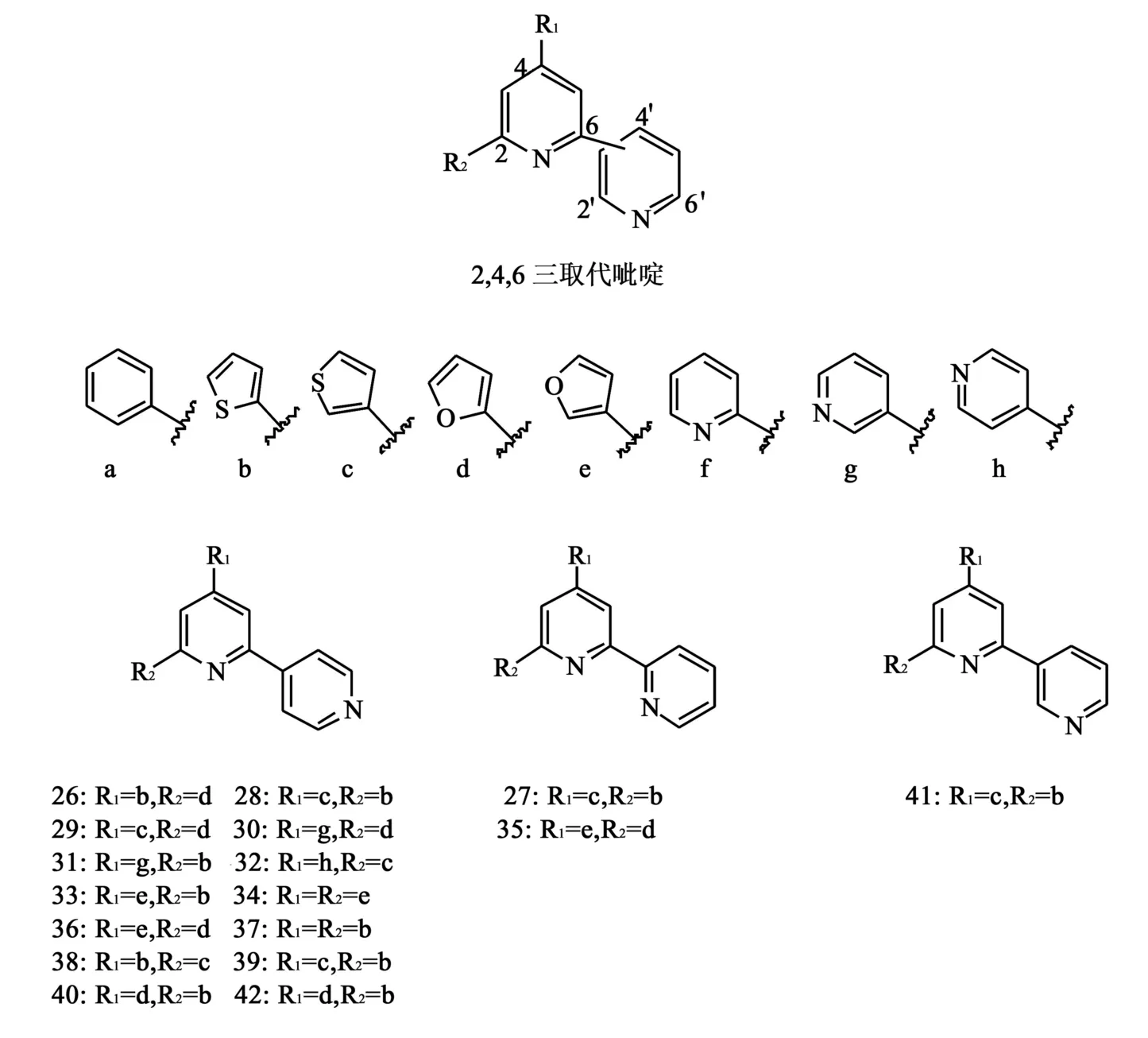
图10 2,4,6三取代吡啶类似物结构
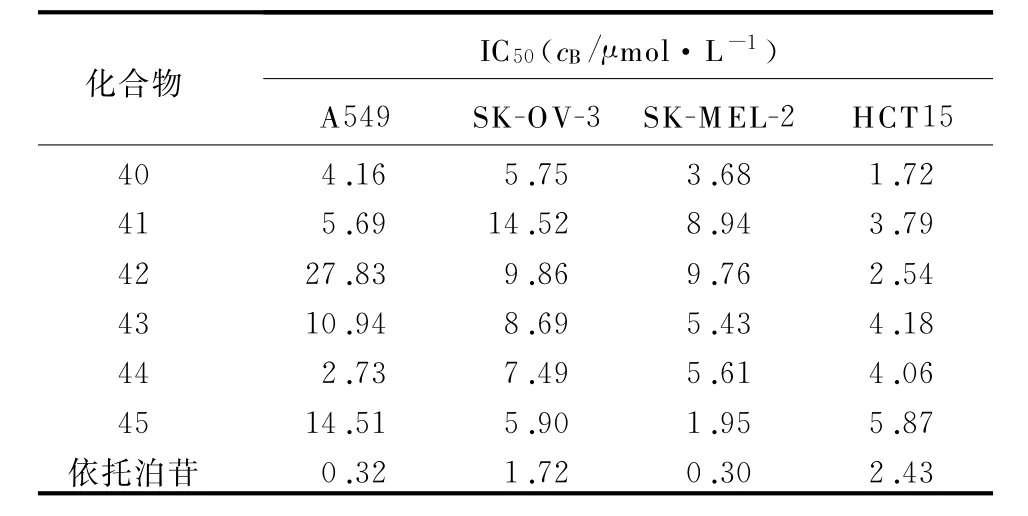
表1 化合物40~45对人4种肿瘤细胞的检测结果
1.11 Benzothiopyranoindoles 2009年,Dalla等[29]发现四环化合物benzothiopyranoindoles类似物具有抑制肿瘤增殖的活性。通过分子对接等手段最终发现,吲哚环部分与N原子直接相连的N-甲基乙胺基侧链可以通过质子化与DNA分子上富含AT碱基对的区域以离子静电力作用结合,嵌入在DNA分子的小沟中。在人宫颈癌细胞和HL-60细胞株生长抑制实验中,该类似物(图11)的IC50值为0.45~3.21μmol/L。此外,还证实该类化合物的抗肿瘤活性确与N-甲基乙胺基侧链有关,且不受生色团影响。该类化合物在较低剂量下主要通过抑制TopⅡ活性引发细胞毒作用,而在更高浓度时可以通过对TopⅠ和TopⅡ的双重抑制,抑制肿瘤细胞的生长。

图11 benzothiopyranoindoles类似物结构
2 讨论
TopⅠ和TopⅡ的单一抑制剂是现在临床上使用较多的广谱抗肿瘤化疗药物,但由于肿瘤对该类药物普遍具有耐药性,从而导致临床治疗效果降低。而近几年Tops双重抑制剂的诞生使得该类药物的临床疗效大幅提升。本综述通过总结近年TopⅠ/TopⅡ双重抑制剂的相关报道,意在归纳双分子靶标抗肿瘤药物的进展。以上药物主要分为3类:①研究较为成熟的药物。tafluposide、batracylin、吩嗪类衍生物XR11576和XR5944是现在已经报道较多的、抑制机制较为明确的TopⅠ/TopⅡ双重抑制剂,也是很有发展潜力的候选药物。②植物提取的天然产物及其衍生物7~24。这些天然及其衍生物虽然往往不止是一种结构,但它们却拥有相似的作用机制,这类化合物可以通过结构改造来改善药效。③人工合成化合物25~43。目前主要通过基团转换研究构效关系,并筛选得到拥有双靶标机制的抗肿瘤药物。其中双环衍生物和BN80927被认为是活性较为良好的候选药物。
TopⅠ/TopⅡ双重抑制剂在未来的抗肿瘤药物研究中有较为广阔的市场,这些TopⅠ/TopⅡ双重抑制剂如tafluposide、XR11576、BN80927在降低药物毒性、克服耐药性、提高药物耐受性和有效性等方面迈出重要一步,取得了长足的发展,有些药物已经进入临床Ⅲ期研究,即将上市。未来研究可着眼于通过研究不同结构的药物对TopⅠ和TopⅡ作用的相似机制来诠释药物结构与TopⅠ/TopⅡ双靶的作用关系,为设计出更为合理的TopⅠ/TopⅡ双重抑制剂,开发高效低毒的双靶标抗肿瘤药物服务。
[1] Wall ME,WaniMC,Cook CE,etal.Plant antitumor agentsⅠ.The isolation and structure of cam ptothecin,a noveⅠalkaloidal leukem ia and tumor inhibitor from cam ptotheca acum inata1,2[J].JAm Chem Soc,1966,88(16):3888-3890.
[2] Hsiang YH,Hertzberg R,Hecht S,et al.Camptothecin induces protein-linked DNA breaks viamammalian DNA topoisomeraseⅠ[J].JBiol Chem,1985,260(27):14873-14878.
[3] Dallavalle S,Gattinoni S,M azzini S,et al.Synthesis and cytotoxic activity of a new series of topoisomeraseⅠinhibitors[J].Bioorg M ed Chem Lett,2008,18(4):1484-1489.
[4] K ruczynski A,Barret JM,Van H ille B,etal.Decreased nucleotide excision repair activity and alterations of topoisomeraseⅡalpha are associated w ith the in vivo resistance of a P388 leukem ia subline to F11782,a novel cataly tic inhibitor of topoisomerasesⅠandⅡ[J].Clin Cancer Res,2004,10(9):3156(3168.
[5] K luza J,M azinghien R,Irw in H,et al.Relationships between DNA strand breakage and apoptotic progression upon treatment of HL(60 leukem ia cells w ith tafluposide or etoposide[J].Anticancer Drugs,2006,17(2):155(164.
[6] M ucci(LoRusso P,Polin L,Bissery MC,et al.Activity of batracylin(NSC-320846)against solid tumors of m ice[J].Invest New Drugs,1989,7(4):295-306.
[7] Plowman J,Paull KD,A tassi G,et al.Preclinical antitumor activity of batracylin(NSC 320846)[J].Invest New Drugs,1988,6(3):147-153.
[8] Rao VA,Agama K,Holbeck S,et al.Batracylin(NSC 320846),a dual inhibitor of DNA topoisomerasesⅠandⅡinduces histone gamma-H 2AX as a biomarker of DNA damage[J].Cancer Res,2007,67(20):9971-9979.
[9] Lew is LJ,M istry P,Charlton PA,et al.M ode of action of the novel phenazine anticancer agents XR 11576 and XR5944.Anticancer D rugs 2007,18,139-48.
[10] de Jonge M J,Kaye S,Verw eij J,etal.Phase Iand pharmacokinetic study of XR11576,an oral topoisomeraseⅠandⅡinhibitor,adm inistered on days 1-5 of a 3-w eekly cycle in patients w ith advanced solid tumours[J].Br JCancer,2004,91(8):1459-1465.
[11] Verborg W,Thomas H,Bissett D,et al.First-into-man phaseⅠand pharmacokinetic study of XR5944.14,a novel agent w ith a unique mechanism of action[J].Br J Cancer,2007,97(7):844-850.
[12] M ontaner B,Castillo-Avila W,M artinell M,et al.DNA interaction and dual topoisomeraseⅠandⅡinhibition properties of the anti-tumor d rug prodigiosin[J].Toxicol Sci,2005,85(2):870-879.
[13] M ontaner B,Navarro S,Pique M,et al.Prodigiosin from the supernatant of Serratia marcescens induces apoptosis in haematopoietic cancer cell lines[J].Br J Pharmacol,2000,131(3):585-593.
[14] M ontaner B,Perez-Tomas R.Prodigiosin-induced apop tosis in human colon cancer cells[J].Life Sci,2001,68(17):2025-2036.
[15] Lin JK.Molecular targets of curcumin[J].Adv Exp Med Biol,2007,595:227-243.
[16] Lopez-Lazaro M,W illmore E,Jobson A,etal.Curcum in induces high levels of topoisomeraseⅠ-andⅡ-DNA comp lexes in K562 leukem ia cells[J].JNat Prod,2007,70(12):1884-1888.
[17] Dhandapani KM,Mahesh VB,Brann DW.Curcumin supp resses grow th and chemoresistance of human glioblastoma cells via AP-Ⅰand NF kappaB transcrip tion factors[J].J Neurochem,2007,102(2):522-538.
[18] Chih LL,Jen KL.Curcumin:a potential cancer chemopreventive agent through supp ressing NF-κB signaling[J].J Cancer Mol,2008,4(1):11-16.
[19] Choi JY,Seo CS,Zheng MS,etal.TopoisomeraseⅠandⅡinhibitory constituents from the bark of Tilia amurensis[J].A rch Pharm Res,2008,31(11):1413-1418.
[20] Ishiyama D,Kanai Y,Senda H,et al.Novel human topoisomeraseⅠinhibitors,topopyrones A,B,C and D.Ⅱ.Structure elucidation[J].JAntibiot(Tokyo),2000,53(9):873-878.
[21] Kanai Y,Ishiyama D,Senda H,et al.Novel human topoisomeraseⅠinhibitors,topopy rones A,B,C and D.Ⅰ.Producing strain,fermentation,isolation,physico-chem ical properties and biological activity.J Antibiot(Tokyo)2000,53,863-72.
[22] Khan QA,Elban MA;Hecht SM.The topopy rones poison human DNA topoisomerasesⅠandⅡ[J].JAm Chem Soc,2008,130(39):12888-12889.
[23] Hecht SM,Khan QA,Maini R,et al.Topopyrones:Dual topoisomerase inhibitors.Patent PCT/US2009/050081,2010.
[24] López-Lázaro M,W illmore E,Austin CA.The dietary flavonoidsmy ricetin and fisetin act as dual inhibitors of DNA topoisomerasesⅠandⅡin cells[J].M utat Res,2010,696:41-47.
[25] Demarquay D,Huchet M,Coulomb H,et al.BN80927:A novel homocamptothecin that inhibits proliferation of human tumor cells in vitro and in vivo[J].Cancer Res,2004,64:4942-4949.
[26] Lavergne O,Demarquay D,Bailly C,et al.TopoisomeraseⅠ-mediated antiproliferative activity of enantiomerically pure fluorinated homocamptothecins[J].J Med Chem,2000,43(11):2285-2289.
[27] Taniguchi K,Kohno K,Kaw anam i K,et al.D rug-induced down-regulation of topoisomeraseⅠin human epidermoid cancer cells resistant to saintopin and cam ptothecins[J].Cancer Res,1996,56(10):2348-2354.
[28] Basnet A,Thapa P,Karki R,et al.2,4,6-Trisubstituted pyridines:synthesis,topoisomeraseⅠandⅡinhibitory activity,cy totoxicity,and structure-activity relationship[J].Bioorg Med Chem,2007,15(13):4351-4359.
[29] Dalla Via L,M agno SM,Gia O,et al.Benzothiopy ranoindole-based antiproliferative agents:synthesis,cytotoxicity,nucleic acids interaction,and topoisomerases inhibition properties[J].JM ed Chem,2009,52(17):5429-5441.
Research progress of dual topoisomeraseⅠandⅡinhibitors
JIANG Yan,SHENGChunquan,DONGGuoqiang(DepartmentofMedicinalChemistry,Schoolof Pharmacy,Second M ilitary Medical University,Shanghai200433,China)
DNA topoisomerases(Tops)are essential enzymes that regulate the cellular processes such as replication,transcription,recombination and repair.DNA Tops can be classified into two types,topoisomeraseⅠ(TopⅠ)and topoisomeraseⅡ(TopⅡ).They catalyze the breakage and religation of DNA,maintaining the topological changes of DNA and various DNA metabolic processes.Due to their important role in DNA metabolism,the ability to interfere w ith the functions of Tops or generating Top-mediated DNA damage is an effective strategy for cancer chemotherapy.Tops have been considered as themost important targets for tumor chemotherapy.In this review,we used examples to describe the development of dual topoisomeraseⅠandⅡinhibitors.
topoisomeraseⅠ;topoisomeraseⅡ;dual inhibitors;anticancer drugs
R979.1
A
1006-0111(2015)04-0303-07
10.3969/j.issn.1006-0111.2015.04.004
2013-10-15
2014-02-19[本文编辑]李睿旻
蒋 琰,硕士研究生.E-mail:jy930626@sina.com
董国强,讲师.研究方向:抗肿瘤药物化学.E-mail:dgq-81@163.com
