脊髓小脑共济失调2型家系的临床表现和基因型分析
2015-05-08吴士文
孙 宇,吴士文
脊髓小脑共济失调2型家系的临床表现和基因型分析
孙 宇1,2,吴士文2
目的 探讨脊髓小脑共济失调2型(spinocerebellar ataxia 2,SCA2)患者的临床表现和分子遗传学特征。方法 描述一家系4代SCA2部分成员的临床表现,总结其特点,并检测基因分型。结果 该家系4代共15例患者发病,均符合常染色体显性遗传特征,14例(除先证者的外婆发病情况不详外)均以步态不稳为首发症状,伴不同程度构音障碍,均有明显的腱反射减弱、四肢远端肌肉萎缩,部分患者伴慢眼动、动作性震颤、不自主摇头、癫痫等。其中第1代患者发病年龄不详,第2代患者于50岁左右发病,第3代患者于36~47岁发病,第4代患者于21岁发病,发病年龄呈逐代提前现象。先证者及其同胞兄弟患者SCA2/ATXN2基因(CAG)n重复数目为39次,其侄子(CAG)n重复数目为50次,发病明显提前且症状重。结论 SCA2家系中存在遗传早现现象,除小脑性共济失调外,还可伴有其他特征性表现。这些临床特征有助于诊断与鉴别诊断,但明确诊断依靠基因学检测。
脊髓小脑共济失调;基因型分析;临床特征
SCA是一组包含多种共济失调亚型的、具有高度临床和遗传异质性的神经系统退行性疾病[1]。目前,普遍认可的致病机制是致病基因编码区内CAG三核苷酸重复序列异常扩增导致编码蛋白内形成异常扩展的多聚谷氨酸肽链,引起编码蛋白的错误折叠,在中枢神经系统-小脑等部位的神经元内形成泛素阳性核内包涵体,并产生选择性细胞毒性作用[2]。除了编码区CAG重复序列异常扩增是SCA发病的原因外,编码区和非编码区碱基的缺失、错义突变及剪切位点突变均有可能导致SCA。目前已经报道有30余种亚型,各亚型的临床表现相似,交替重叠,但又各具特点[3]。在我国最常见的是SCA3,其次是SCA2和SCA6[4]。2013年武警总医院收录了一家系4代15例SCA2患者的病例资料,笔者对于该家系部分患者的临床表型和基因型进行了分析,旨在为SCA2的临床诊断与鉴别诊断提供依据。
1 对象与方法
1.1 对象 本组SCA2患者来自山西晋中,于武警总医院神经内科就诊,其家系四代共15例患者(图1),汉族,均符合常染色体显性遗传特点且临床诊断符合Harding标准。
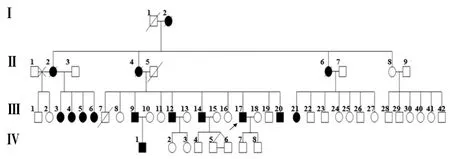
图1 SCA家系图谱
先证者:Ⅲ-17,男,43岁。2013-10因“进行性行走不稳6年余”首次就诊于武警总医院,之后长期随访。患者于2007年无明显诱因出现行走不稳,表现为步行缓慢,步距宽。2年后发现四肢远端肌肉明显萎缩,以双手大小鱼际明显,精细活动完成不佳。近2~3年,患者自觉上述症状加重,出现蹲下起立困难、行走直线不能、持物不稳,并逐渐出现言语不清。家族史如图1所示。神经系统查体:神志清,构音障碍。双侧瞳孔等大等圆,直径约3 mm,对光反射灵敏,可疑突眼,轻度慢眼动。四肢远端肌力4级,近端肌力5级;腱反射减弱,肌张力正常。双侧指鼻试验、跟膝胫试验欠稳准,双侧病理征阴性。动作性震颤,宽基步态,Romberg征阳性。
患者Ⅰ-2,女,已故。为患者外婆。逝去前曾有行走不稳、肌肉萎缩、言语不清等类似症状。具体发病时间不详。
患者Ⅱ-2、Ⅱ-6,均为女性,已故。二者为患者姨母,皆于50岁左右起病,表现为言语笨拙、行走不稳、肌肉萎缩,最终瘫痪,因并发症于65岁左右去世。
患者Ⅱ-4,女,已故。为患者母亲,51岁起病,表现为行走不稳,言语笨拙,58岁时明显加重,63岁之后卧床。67岁因行动障碍摔伤导致骨盆骨折死亡。
患者Ⅲ-3、Ⅲ-4、Ⅲ-5、Ⅲ-6、Ⅲ-21,均为患者表姐,皆于40岁左右发病,表现为行走不稳、言语不清、肌肉萎缩,并随病程进展均出现动作性震颤、突眼、眼球扫视速度变慢等症状。目前Ⅲ-3、Ⅲ-4瘫痪在床,生活不能自理,伴有饮水呛咳,Ⅲ-3、Ⅲ-4、Ⅲ-5伴头部不自主晃动,Ⅲ-21伴癫痫病史。
患者Ⅲ-9,男,59岁,为患者二哥,44岁发病,最先出现身体摇晃、行走不稳,之后出现言语笨拙、饮水呛咳,55岁时难以独自站立,行走需辅助工具。近4年不能行走,小便失禁。查体:神志清,重度构音障碍,头部不自主晃动,眼睑退缩,双眼球固定。四肢远端肌肉萎缩严重,双上肢近端肌力3级、远端肌力3级,动作性震颤。肌张力减低,四肢腱反射消失,病理征未引出。指鼻及跟膝胫试验不能完成。
患者Ⅲ-12,男,55岁,为患者三哥,47岁发病,目前行走不稳,轻度构音障碍,双上肢远端肌肉萎缩较重,双手伸直困难,持物不稳,轻度动作性震颤。
患者Ⅲ-14,男,46岁,为患者四哥,40岁发病,目前症状与先证者相似。
患者Ⅲ-20,男,41岁,为患者七弟,36岁发病,目前行走不稳,宽基步态。有癫痫病史。
患者Ⅳ-1,男,25岁,为患者二哥之子,21岁发病,开始出现行走不稳伴持物不稳,言语欠流畅,并逐渐加重,近1年丧失行走能力,生活自理困难。查体:神志清,重度构音障碍,头部不自主摇晃,伴有动作性震颤,突眼,慢眼动。四肢远端肌肉萎缩,双上肢近端肌力4级、远端肌力3级。四肢肌张力降低,腱反射减弱,病理征未引出。指鼻试验欠稳准,轮替笨拙,跟-膝-胫试验不能完成。
1.2 方法
1.2.1 样本采集 依据“知情同意原则”,空腹采集该家系中的5名患者(Ⅲ-9、12、17、20,Ⅳ-1)及5名无SCA表现个体(Ⅲ-8、11、19,Ⅳ-7、8)的外周静脉血5 ml,乙二胺四乙酸(Ethylene Diamine Tetraacetic Acid, EDTA)抗凝后采用经典酚-氯仿抽提法提取基因组DNA作为聚合酶链反应(polymerase chain reaction, PCR)模板。
1.2.2 SCA2基因CAG三核苷酸重复突变检测 根据SCA2基因第1外显子CAG重复片段两侧序列,由北京康旭基因检测公司设计引物序列,正向引物(F引物)序列:5’-GGGCCCCTCACCATGTCG-3’;反向引物(R引物)序列:5’-CGGGCTTGCGGACATTGG-3’。PCR扩增体系(25 μl)包括:2×GC Buffer Ⅰ 12.5 μl,dNTP 4 μl,LA Taq 0.25 μl,Primer(FP荧光引物) 2 μl,模板DNA2.0 μl,余下体积由灭菌双蒸水补足。扩增条件:94 ℃预变性1 min,94 ℃变性30 s,60 ℃退火30 s,72 ℃延伸30 s,运行30个循环;72 ℃终延伸1 h。在PTC200PCR反应仪中进行PCR扩增,2%琼脂糖凝胶电泳鉴定,使用3130基因测序仪进行测序分析。根据已知片段大小对应的CAG重复次数的关系(116 bp约重复22次),计算样本对应的的CAG重复次数。
2 结 果
2.1 发病特点及临床体征 该家系4代共15例患者,其中男6例,女9例,每代都有患者发病,且无明显的性别差异,符合常染色体显性遗传规律。第1代患者发病年龄不详,第2代患者50岁左右发病,65~70岁去世,病程15年左右。第3代患者36~47岁发病,病程5~15年,其中3例于发病10年后不能独立行走。第4代患者21岁发病,病程4年,已丧失行走能力。
14例(除先证者的外婆发病情况不详外)均以步态不稳为首发症状,伴有不同程度的构音障碍,肌肉萎缩明显,上述症状呈进行性加重。其中4例已经去世,在世的11例患者临床表现形式多样,丧失行走能力4例,构音障碍10例,远端肌肉萎缩10例,动作性震颤10例,头部不自主晃动5例,慢眼动(包括可疑)9例,腱反射减弱10例,癫痫病史2例,肌张力障碍1例,未见明显认知障碍者。
2.2 基因诊断结果 在该家系中,5名无SCA表现个体(Ⅲ-8、11、19,Ⅳ-7、8)的2条等位基因的PCR产物为116bp,均无CAG三核苷酸过度扩增,重复次数约为22次,与健康人群中正常重复次数相同,与其临床表型相符。
5名患者均有1条等位基因的CAG三核苷酸过度扩增,其中Ⅲ-9、12、17、20的SCA2基因编码区(CAG)n三核苷酸重复数目异常,为22/39次;Ⅳ-1的SCA2基因编码区(CAG)n三核苷酸重复数目异常,为22/50次。见图2。
3 讨 论
SCA2最初是在古巴的奥尔金省被发现并描述的,致病基因为ATXN2[5],由Gispert等[6]在1993年定位于12q24.13,其CAG重复位于基因编码区第一外显子。目前得到广泛认可的观点是:SCA2基因CAG三核苷酸正常重复范围为13~31次,异常重复范围>34次,介于32~34次者为中间重复范围。根据文献[5,7]报道,重复次数>31次者即可出现神经退行性改变的临床症状。临床上,SCA2可以体现为小脑综合征或帕金森综合征,主要表现为进行性的共济失调、构音障碍,而后期病变则可累及脑干、脊髓、丘脑等。这种特殊的萎缩模式类似于多系统萎缩(multiple system atrophy,MSA),因此还可能伴有扫视缓慢、腱反射减弱、位置或动作性震颤、肌阵挛等特征性体征,也可伴随锥体外系症状及认知障碍[8]。
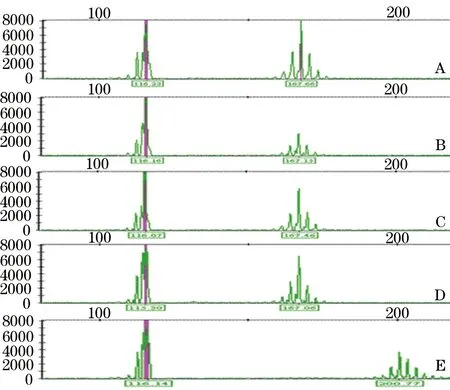
图2 DNA片段分析
该家系四代患者之间发病年龄跨度很大,文献[9]报道指出SCA1、2、3、7亚型间的发病年龄差异无统计学意义,吴英等[10]也在其临床试验中验证了这一结论,因此发病年龄对于SCA的分型诊断意义不大。因第1、2代患者均已经去世,无法获得临床资料及基因检测结果。第3代患者CAG重复次数为39次,第4代患者重复次数为50次,均在异常范围内。第4代患者发病年龄较第3代明显提前,病程仅4年即丧失行走能力,且出现较多伴发症状,这符合SCA2的遗传早现现象[11],Tezenas等[12]研究发现发病年龄主要受CAG重复扩展所调控,SCA2的发病年龄与CAG重复次数呈负相关关系,严重程度及进展速度与CAG重复次数呈正相关关系。
该家系中的先证者及多名患者均存在明显的步态不稳、构音障碍等症状,同时伴有慢眼动、腱反射减弱、动作性震颤、肌肉萎缩等特征性表现,这与文献[13]报道的SCA2常见表现一致。既往研究报道中5%~19%的SCA2患者存在不同程度的认知障碍[14],Ma等[15]认为SCA2的认知障碍与疾病严重程度相关,而与年龄、发病年龄、受教育年限、疾病持续时间等无关,但本研究报道的家系未见明显认知障碍者。与其他SCA亚型相比,SCA2具有慢眼动,即眼球扫视速度过低这一特征性表现,在该家系中9例症状严重患者出现慢眼动症状,而2例病程较短、症状较轻患者则未出现眼部异常体征,再次证明了只有当SCA2疾病进展到一定阶段时才会出现慢眼动体征,该体征在临床诊断与分型中有重要意义,同时也是疾病严重程度的标志[16]。在该家系中多人出现了行走缓慢、震颤、头部不自主晃动等帕金森综合征症状,一项针对中国人群的研究证实ATXN2 CAG扩张是导致脊髓小脑共济失调2型-帕金森综合征(spinocerebellar ataxia 2-Parkinson’s disease,SCA2-P)的唯一原因[17]。另外在该家系中的2例患者既往有癫痫病史,癫痫病史以前很少与SCA2相关联,Tan等[18]曾对此提出了单独的链接癫痫易感性基因扩展SCA2基因引起相互作用的假设,虽然尚未得到验证,但仍具有一定的意义。
据以上分析,在一个家系中存在构音障碍、行走不稳、远端肌肉萎缩、动作性震颤、头部不自主晃动、腱反射减弱、慢眼动、癫痫病史等症状的患者,且发病年龄具有逐代提前的特点时,诊断应考虑SCA2,但明确诊断依赖于基因检测。
[1] Schols L, Bauer P, Schmidt T,etal.Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis[J].Lancet Neurol,2004,3(5):291-304.
[2] Metilla-Duenas A, Ashizawa T, Brice A,etal.Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias[J].Cerebellum,2014,13(2):269-302.
[3] Shakkottai V G, Fogel B L.Clinical neurogenetics autosomal dominant Spinocerebellar ataxia[J].Neurol Clin,2013,31(4):987-1007.
[4] Tang B S, Liu C Y, Shen L,etal.Frequency of SCA1, SCA2, SCA/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds[J].Arch Neurol,2000,57(4):540-544.
[5] Pulst S M, Santos N, Wang D,etal.Spinocerebellar ataxia type 2: polyQ repeat variation in the CACNA1A calcium channel modifies age of onset[J].Brain,2005,128(10):2297-2303.
[6] Gispert S, Twells R,Orozco G,etal.Chromosomal assignment of the second locus for autosomal dominant cerebellar ataxia (SCA2) to chromosome 12q23-24.1[J].Nat Genet,1993,4(3):295-299.
[7] Pulst S M,Nechiporuk A, Nechiporuk T,etal.Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2[J].Nat Genet,1996,14(3):269-276.
[8] Lastres-Becker I, Rub U, Auburger G.Spinocerebellar ataxia type 2 (SCA2) [J].Cerebellum,2008,7(2):115-124.
[9] van de Warrenburg B P, Sinke R J, Verschuuren-Bemelmans C C,etal. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis[J]. Neurology, 2002,58(5):702-708.
[10] 吴 英,魏倩倩,商慧芳.脊髓小脑共济失调基因型分布及临床特点分析[J].中国实用内科杂志,2014,34(5):512-515.
[11] Choudhry S, Mukerji M, Srivastava A K,etal.CAG repeat instability at SCA2 locus: anchoring CAA interruptions and linked single nucleotide polymorphisms[J].Hum Mol Genet,2001,10(21):2437-2446.
[12] Tezenas du Montcels, Durr A, Bauer P,etal.Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes[J].Brain,2014,137(9):2444-2455.
[13] 宋 旸,陈 彪,温 玫.脊髓小脑共济失调1、2、3型的临床表现和基因分型[J].疑难病杂志,2006,5(1):19-22.
[14] Bürk K,Stevanin G,Didierjean O,etal.Clinical and genetic analysis of three German kindreds with autosomal dominant cerebellar ataxia type 1 linked to the SCA2 locus[J].J Neurol,1997,244(4):256-261.
[15] Ma J, Wu C, Lei J,etal.Cognitive impairments in patients with spinocerebellar ataxia types 1, 2 and 3 are positively correlated to the clinical severity of ataxia symptoms[J].Int J Clin Exp Med,2014,7(12):5765-5771.
[16] Seifried C,Velazquez-Perez L,Santos-Falcon N,etal.Saccade velocity as a surrogate disease marker in spinocerebellar ataxia type 2[J].Ann N Y Acad Sci,2005(1039):524-527.
[17] Wang C, Xu Y, Feng X,etal.Linkage analysis and whole-exome sequencing exclude extra mutations responsible for the parkinsonian phenotype of spinocerebellar ataxia-2[J].Neurobiol Aging,2015,36(1):545.e1-e7.
[18] Tan N C, Zhou Y, Tan A S,etal.Spinocerebellar ataxia type 2 with focal epilepsy--an unusual association[J].Ann Acad Med Singapore,2004,33(1):103-106.
(2015-03-04收稿 2015-03-27修回)
(责任编辑 罗发菊)
Analysis on clinical manifestations and genotyping in a large SCA2 pedigree
SUNYu1,2andWUShiwen2.
1.ClinicalCollegeofAnhuiMedicalUniversity,2.DepartmentofNeurology,GeneralHospitalofChinesePeople’sArmedPoliceForces,Beijing100039,China
WUShiwen,Email:neurowu@gmail.com
Objective To explore the clinical features and genetic characteristics in spinocerebellar ataxia 2(SCA2) patients.Methods Clinical manifestations of some patients’ pedigrees were recorded and summarized, while their genotyping were analyzed. Results There were 15 patients in this family with autosomal dominant inheritance feature. Except the condition of maternal grandmother was unknown, initial symptoms in the other 14 affected members were gait disorders accompanied by dysphasia, hyporeflexia, amyotrophy, and some members with slow eye movements, action tremor, involuntary nodding, and epilepsy. Onset age became earlier from generation to generation in the family which was around 50 year-old, 36 to 47 year-old, 21 year-old in generation Ⅱ, Ⅲ, Ⅳ, respectively. The number of CAG repeats in SCA2/ATXN2 allele were 39 in proband, as well as in his brothers with symptoms, while 50 in one nephew of proband with more serious clinical symptoms. Conclusions Genetic anticipation is found in this family with SCA2. Cerebellar ataxia is noted in most patients accompanied by other different symptoms and signs. Clinical features are helpful in the diagnosis and differential diagnosis of SCA2, while definitive diagnosis depends on genetic testing.
spinocerebellar ataxias; genetic analysis; clinical features
10.13919/j.issn.2095-6274.2015.04.007
孙 宇,硕士研究生在读,E-mail:sdcysunyu@126.com
100039 北京,武警总医院:1.安徽医科大学北京武警总医院临床学院,2.神经内科
吴士文,E-mail:neurowu@gmail.com
R744;R394
