一株产谷氨酰胺转氨酶菌株的分离、鉴定及其tgl的克隆表达
2015-05-05张莹莹杜萍萍申培立李志辉于宏伟卢海强苏旭东檀建新
张莹莹,石 楠,2,杜萍萍,申培立,李志辉,于宏伟,卢海强,苏旭东,张 伟,檀建新,*
(1.河北农业大学食品科技学院,河北省农产品加工工程技术中心,河北保定 071001;2.河北大学生命科学学院,河北保定 071002)
一株产谷氨酰胺转氨酶菌株的分离、鉴定及其tgl的克隆表达
张莹莹1,石 楠1,2,杜萍萍1,申培立1,李志辉1,于宏伟1,卢海强1,苏旭东1,张 伟1,檀建新1,*
(1.河北农业大学食品科技学院,河北省农产品加工工程技术中心,河北保定 071001;2.河北大学生命科学学院,河北保定 071002)
用稀释平板法从土壤样品中分离到166株细菌菌株,通过凝胶法初筛和Folk比色法复筛,得到一株产谷氨酰胺转氨酶(transglutaminase,TGase)的菌株,通过形态学、生理生化特征和16S rDNA序列比对证明该菌株是枯草芽孢杆菌(Bacillussubtilis),命名为TGase1318。克隆了该菌株TGase的编码基因tgl,序列长738bp,编码由245个氨基酸组成的蛋白质;TGase的氨基酸序列与NCBI公布的B.subtilis的TGase相似性达94%~100%。用B.subtilis表达载体pTZ和E.coli表达载体pET21b分别构建了含tgl的重组质粒,转化B.subtilisWB800和E.coliBL21,tgl基因表达产物在70℃下可催化BSA交联,表明tgl在B.subtilis和E.coli中获得了表达且表现出TGase活性,这为其在食品工业上的开发利用打下基础。
谷氨酰胺转氨酶,枯草芽孢杆菌,克隆和表达,16S rDNA
谷氨酰胺转胺酶(Transglutaminase,简称TGase,EC2. 3. 2. 13)是一种催化酰基转移反应的酶。它以肽链中谷氨酰胺残基的γ-羧酰胺基作为酰基供体,酰基受体可以是多肽链中赖氨酸残基的ε-氨基、伯胺基或水。通过催化食品中的蛋白质的交联反应,从而改善其凝胶性、黏度、乳化性等物理性质,在食品加工过程中有广泛应用[1]。谷氨酰胺转胺酶存在于动物、植物和微生物中,相对于动、植物来源的TGase的分离、提取、纯化工艺复杂、回收率低、成本过高等缺点,微生物源的TGase可通过发酵法制备,具有产量高、易回收、成本低、不受季节限制等优点,更适合工业化生产和应用[2]。
利用生物技术将编码TGase的基因克隆并在适当受体微生物中表达,是提高TGase产量的有效途径。目前,微生物谷氨酰胺转胺酶(Microbial transglutaminase,MTG)表达的研究主要集中在对放线菌的tgl基因的克隆与表达上[3-5]。Kikuchi[6]等人利用Corynebacterium glutamicum构建了分泌型表达质粒,可以直接表达具有酶活性的TGase,通过优化发酵条件,使产量达到142mg/mL。有关枯草芽孢杆菌TGase的报道不少,但主要集中于TGase在芽孢形成时促进孢衣蛋白之间的交联作用[7-8],而对tgl基因克隆和表达则鲜有报道[9]。鉴于芽孢的抗热性,其TGase可能有较高的最适温度,较放线菌来源的TGase可能更适合高温条件下的食品加工。如果能够通过基因工程的手段获得高效表达源于Bacillus的TGase的重组菌株,则其将会在食品工业中起到重要的作用。
本研究从土壤中分离并筛选出产TGase的菌株,克隆了其tgl基因,导入了枯草芽孢杆菌和大肠杆菌进行表达,为提高TGase表达量提供理论基础。
1 材料与方法
1.1 材料与仪器
土壤样品 河北省保定地区,取地表5~15cm之间的土壤,装入无菌袋中4℃保存;E.coliBL21 购自宝生物工程(大连)有限公司;B.subtilisWB800,pET21b和pTZ表达载体 为本实验室保存;重组质粒pET21b-tgl,pTZ-tgl和TGase表达菌株BL21-pET1b-tgl,WB800-pTZ-tgl 为本研究构建;试剂CBZ-Gln-Gly、盐酸羟胺、还原性谷胱甘肽、L-谷氨酸-γ-单羟肟酸 均购自Sigma公司,其余试剂均为国产分析纯试剂;质粒提取试剂盒,胶回收试剂盒,pMD19-T载体和限制性内切酶等分子生物学试剂 购自宝生物工程(大连)有限公司;PCR试剂、100bp marker(2000、1000、900、800、700、600、500、400、300、200、100bp)、1kb marker(10、8、6、5、4、3、2、1kb) 购自北京康为世纪生物科技有限公司。
紫外分光光度仪 上海光谱仪器有限公司;JY96-IIN超声波细胞粉碎机 宁波新芝生物科技有限公司;DYY-8C电泳仪 北京六一仪器厂;BINDA 2020D凝胶成像系统 北京宾达英创科技有限公司。
1.2 培养基
Luria-Bertani(LB)培养基:胰蛋白胨10.0g/L、酵母粉5.0g/L、NaCl 10.0g/L、琼脂粉15.0g/L、蒸馏水1000mL,pH8.0。
发酵培养基:蛋白胨20.0g/L,可溶性淀粉20.0g/L、酵母粉2.0g/L、MgSO42.0g/L、KH2PO42.0g/L、K2HPO42.0g/L、蒸馏水1000mL,pH7.0
1.3 实验方法
1.3.1 产酶菌株的分离与筛选
1.3.1.1 细菌的分离、纯化 利用稀释涂布平板法[10]在LB平板上分离土壤样品中的细菌,30℃下培养2~3d,根据菌落形状、大小、颜色等特征进行初步归类,每一类随机挑取单菌落划线纯化,并保存于4℃冰箱中备用。
1.3.1.2 初筛 将分离的菌种分别于LB液体培养基中活化后,转接至含5mL发酵培养基的试管中,于30℃、200r/min培养6d后利用凝胶法初筛产酶菌株[11]。
1.3.1.3 复筛 对初筛选出的菌株用比色法进行复筛,测定上清液的谷氨酰胺转胺酶活力。谷氨酰胺转胺酶活力测定按照Folk的比色法[12]。终止反应后,12000r/min离心,弃去沉淀,测上清夜在 525nm下的吸光度。一个MTG酶活单位(1U/mL)定义为37℃下1min催化1μmol底物CBZ-Gln-Gly生成其单羟肟酸产物所需的酶量。
1.3.2 菌株的鉴定
1.3.2.1 形态学观察和生理生化实验 菌株鉴定参照《常见细菌系统鉴定手册》[13]和《伯杰细菌鉴定手册》[14]进行。
1.3.2.2 16S rDNA序列系统进化树 按文献[15]提取菌株TGase1318基因组作为模板,用16S rDNA通用引物5′-GAGAGTTTGATCCTGGCTCAG-3′/5′-AAGGAGGTGATCCAGCCGCA-3′扩增16S rDNA。反应体系为:2μL模板DNA,上下游引物各1μL,2×EsTaq MasterMix 10μL,H2O 6μL。PCR反应条件:94℃、预变性10min,94℃变性45s、55℃退火45s、72℃延伸90s,30个循环,72℃延伸5min。PCR产物经胶回收、连接、转化、质粒提取验证后由北京六合华大基因科技股份有限公司测序。将菌株TGase1318的16S rDNA序列经GenBank的BLAST检索进行系统发育分析,选取属内同源性较高的细菌的16S rDNA序列进行遗传距离计算,采用MEGA软件的Neighbor-Joining模型和BioEdit软件分析遗传距离并绘制系统发育树。
1.3.3Tgl的克隆、测序和序列比对
1.3.3.1Tgl的PCR扩增和克隆 依据NCBI上已公布B.subtilis的TGase基因的起始序列,用DNAMAN设计引物,上游引物Tgl-5:5′-GCGGTCGAC ATGATTATTGTATCAGGAC-3′(下划线为Sal I酶切位点);Tgl-3:5′-TAAAGCTTTTAGCGGACG ATGCGGAAAAG-3′(下划线为Hind Ⅲ 酶切位点)。扩增反应体系为60μL:基因组DNA 2μL,引物(10μmol/L)各3μL,2×EsTaq MasterMix 30μL,ddH2O 22μL。PCR反应条件:94℃、预变性10min,94℃变性40s,58℃退火40s,72℃延伸1min,25个循环,72℃终延伸10min。将PCR产物克隆入p-MD-19T的SalI和Hind Ⅲ位点之间,测序。
1.3.3.2 蛋白序列分析 利用NCBI BLAST、DNAMAN软件分析TGase1318氨基酸序列。利用欧洲分子生物学开放软件包EMBOSS(European Molecular Biology Open Software Suite),在线调用pepstats后,输入TGase1318的TGase氨基酸序列,得到其一级结构和一些基本参数。用AntheProt软件分析蛋白的等电点和蛋白二级结构,分别预测其α-螺旋、β-折叠、β-转角、无规则卷曲的百分含量。利用Phyre2软件对该菌株TGase进行蛋白的保守域和三级结构预测。
1.3.3.3 重组质粒pTZ-tgl和pET-21b-tgl的构建 重组质粒pTZ-tgl和pET21b-tgl的构建步骤如图1所示,用SalI和HindⅢ 双酶切1.3.3.1中的tglPCR产物和载体pTZ、pET21b,经酶切胶回收后的tgl基因片段分别与载体pTZ和pET21b连接,转化E.coliJM109。将验证正确的重组质粒命名为pTZ-tgl和pET21b-tgl。
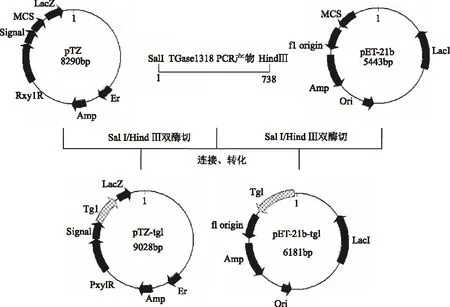
图1 表达载体pTZ-tgl和pET-21b-tgl的构建流程简图Fig.1 Flow chart of the construction of expression vectors pET-21b-tgl and pTZ-tgl
1.3.3.4 重组质粒在E.coliBL21和B.subtilisWB800中的转化 取重组质粒pTZ-tgl加入已制备好的B.subtilisWB800的感受态细胞[16-17]中,至终浓度约1μg/mL,37℃静置1h后200r/min振荡培养3h,转化液涂布在含有红霉素(10μg/mL)LB平板上,培养过夜。挑取阳性转化子验证后命名为WB800-pTZ-tgl。
将重组质粒pET-21b-tgl用热击法转化E. coli BL21感受态细胞,涂布于含100μg/mL的氨苄青霉素LB平板上,经37℃培养,挑取阳性转化子验证后命名为BL21-pET21b-tgl。
1.3.4Tgl在E.coli和B.subtilis中的表达和BSA交联反应 将WB800-pTZ-tgl和BL21-pET21b-tgl分别转接至20mL含有红霉素(10μg/mL)或氨苄青霉素(100μg/mL)的LB液体培养基中,37℃、200r/min震荡培养到OD600为0.5时,分别加入终浓度为2%的D-Xylose和1mmol/L异丙基-β-D-硫代半乳糖苷(IPTG),诱导24h后收集菌体。
将诱导后的WB800-pTZ-tgl发酵液离心收集上清作为粗酶液,参照1.3.1.3方法测定谷氨酰胺转氨酶的活性;同时将60μL粗酶液与30μL BSA(1mg/mL)[18]于70℃保温不同时间,取30μL进行SDS-PAGE电泳分析。
将培养的BL21-pET21b-tgl用IPTG(1mmol/L)诱导后收集菌体,重悬于1mL Tris-HCl(20μmol/L,pH8.0)缓冲液中超声破碎制备成粗酶液,取400μL粗酶液按1.3.1.3测定TGase的活性;取20μL粗酶液进行SDS-PAGE电泳来判定表达产物的大小和多少;同时取60μL粗酶液与30μL BSA(1mg/mL)在70℃保温不同时间,取20μL进行SDS-PAGE电泳分析。以未经IPTG诱导的BL21-pET21b-tgl菌裂解液作为对照。
2 结果与分析
2.1 菌株TGase1318的分离和鉴定
2.1.1 产TGase的芽孢杆菌的分离 经稀释平板法从土样中分离到166株细菌,用酪蛋白凝胶反应初筛得到55株细菌。将这些菌株经摇瓶培养用比色法测定TGase的活性,筛选到一株酶活为0.11±0.04U/mL的菌株,与其他菌株相比,其活性最高,命名为TGase1318,以此为出发菌株作进一步鉴定和异源表达分析。
2.1.2 菌株TGase1318的16S rDNA扩增和系统发育树的构建 以产酶菌株TGase1318的基因组DNA为模板扩增16S rDNA得到长度约为1.5kb的DNA片段,1%琼脂糖凝胶电泳结果见图2。将16S rDNA扩增片段测序,证明该片段长1487bp。将该序列与多个NCBI公布的芽孢杆菌属的菌株16S rDNA序列进行比对,用MEGA软件构建了基于16S rDNA序列的系统发育树(图2),由图可知其16S rDNA序列与枯草芽孢杆菌(B. subtilis)的同源性高达99%以上,证明菌株TGase1318为B.subtilis。

图2 菌株TGase1318的16S rDNA的 PCR产物及其系统发育树Fig.2 16S rDNA PCR product of the strain TGase1318 and its phylogenetic tree注:M:1kb Marker;1:16S rDNA PCR 产物。
2.1.3 菌株TGase1318的形态及生理生化特征 将菌株TGase1318在LB平板上培养,菌落呈圆形,乌白色,表面干燥,粗糙不透明,边缘不规则(图3)。挑取单个菌落镜检,该菌株为革兰氏阳性、直杆状细菌,在营养条件贫乏或生长条件不利的情况下形成芽孢(图3,箭头所指为芽孢)。将菌株TGase进行生理生化鉴定,结果见表1。从菌落特征、个体形态和生理生化鉴定结果可知,该菌株为枯草芽孢杆菌,与16S rDNA结果一致,因此确定菌株TGase1318为B.subtilis。
2.2 菌株TGase1318tgl基因的克隆和表达
2.2.1tgl基因的克隆 通过己知B.subtilis的tgl序列设计引物,以TGase1318基因组DNA为模板进行PCR扩增,经过扩增后得到DNA条带大小约740bp,PCR产物回收后用pMD-19克隆(图4)。将tgl测序证明该基因编码区全长738bp,与GenBank中已公布的B.subtilis的tgl基因序列比对,同源性在93%~99%之间,该基因已在GenBank中注册(Accession number:1742195)。

表1 TGase1318的生理生化特征Table 1 Physiological and biochemical characteristics of the strain TGase1318
注:+:阳性;-:阴性。

图3 TGase1318在LB平板上的菌落形态、菌体及芽孢形态Fig.3 The morphology of cell, spore and colony of the strain TGase1318 on LB plate The arrow point to the spore of TGase1318 strain
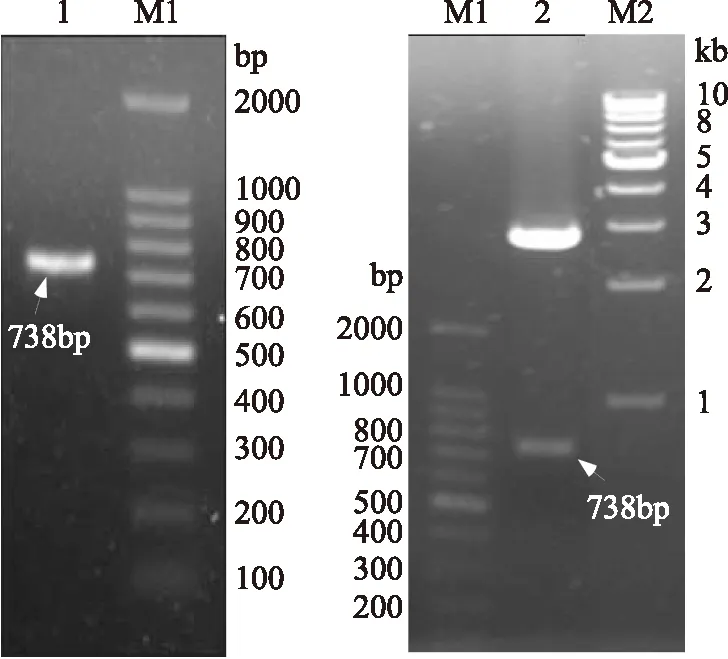
图4 菌株TGase1318的tgl基因的克隆Fig.4 cloning of tgl gene from TGase1318 strain注:1:tgl基因PCR产物; 2:经Sal I/Hind III酶切后的 pMD-19T-tgl;M1:100bp Marker;M2:1kb Marker。
2.2.2tgl编码蛋白质氨基酸序列及特性分析 由tgl基因序列推导出其编码的TGase由245个氨基酸残基组成,包括了20种常见的氨基酸,其中亮氨酸、异亮氨酸和丙氨酸含量最高,分别占11.43%、8.98%、7.76%。从氨基酸性质看,该蛋白含57.55%的非极性氨基酸和42.45%的极性氨基酸,其中极性氨基酸中碱性氨基酸占13.47%,酸性氨基酸占11.43%。该蛋白分子量为28.29ku,预测的等电点为pI 6.71,为弱酸性蛋白。
根据氨基酸序列,基于GOR法预测的蛋白质二级结构见图5,其中α-螺旋、β-折叠、β-转角和无规则卷曲分别占44%、14%、21%和21%,利用Phyre2软件预测的TGase三级结构见图6,它有四个α-螺旋、两个β-折叠,蛋白的保守区预测在蛋白质的中后段,位于第110~197个氨基酸残基区域。将氨基酸序列与NCBI公布的TGase进行BLAST比对,表明该蛋白与现有B.subtilis的TGase同源性高达94%~100%,说明B.subtilis的TGase具有极强的保守性;与其他芽孢杆菌属的TGase相比,同源性多在54%~75%之间,如与B.vallismortis、B.amyloliquefaciens、B.pumilus同源性分别为91%、72%和54%。

图5 tgl编码蛋白TGase的二级结构预测图Fig.5 The secondary structure prediction of TGase encoded by tgl gene注:1:β-折叠;2:β-转角;3:α-螺旋;4:无规则卷曲。

图6 tgl编码蛋白TGase的三级结构预测图Fig.6 The tertiary structure prediction of TGase1318 encoded by tgl gene
2.2.3 含tgl基因的表达载体pTZ-tgl和pET21b-tgl的构建 克隆的tgl基因和分泌型穿梭表达载体pTZ、表达载体pET21b分别经SalI和Hind Ⅲ双酶切,回收后的tgl和载体连接后分别构建重组质粒pTZ-tgl和pET21b-tgl(图1),酶切分析结果证明两个重组质粒含有tgl基因(738bp)和pTZ(约8300bp)(或pET21b,约5440bp)载体,表明重组质粒构建成功(图7)。

图7 含tgl基因表达载体的构建Fig.7 Construction of expression vectors (pET21b-tgl and pTZ-tgl) containing tgl gene注:M1:100bp Marker;M2:1kb Marker 1:经Sal I/Hind II酶切后的pET21b-tgl; 2:经Sal I/Hind III 酶切后的pTZ-tgl。
2.3 表达载体pTZ-tgl和pET21b-tgl在B.subtilisWB800和E. coli BL21中的诱导表达
将含分泌型表达载体pTZ-tgl的阳性菌株WB800-pTZ-tgl在含有红霉素(10μg/mL)的LB培养基中培养至OD600为0.5时,加入木糖诱导24h后离心收集上清,测定谷氨酰胺转氨酶的活性。结果表明TGase酶活较低仅为0.16±0.04U/mL,与出发菌株相似,但是粗酶液与BSA反应不同时间后进行SDS-PAGE电泳分析,可以看出表达的TGase能使BSA交联,并随时间延长交联程度增大(图8),说明该菌株表达且将TGase分泌到了培养基中。但是SDS-PAGE电泳分析发酵液,未发现明显的TGase条带,表明表达量较低。
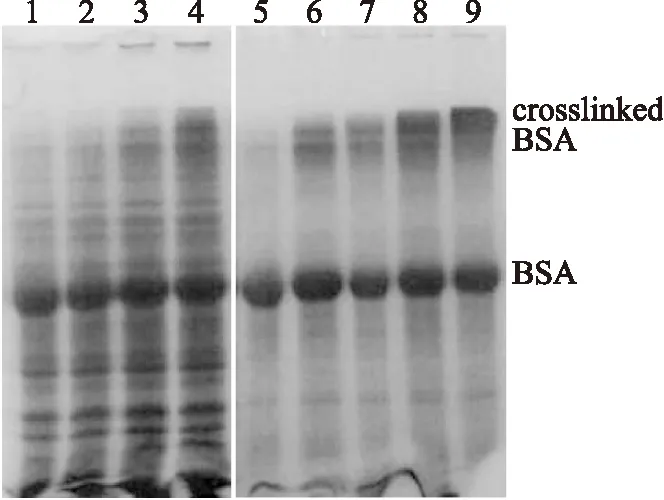
图8 TGase对BSA的交联作用Fig.8 The crosslink of BSA catalyzed by TGase注:1~4:BL21-pET21b-tgl与BSA混合温育0,1,3 and 6h; 5~9:WB800-pTZ-tgl与BSA混合温育0,1,3,6 and 12h。
有鉴于此,我们进而将tgl基因在大肠杆菌中进行了表达,将含pET21b-tgl的阳性菌株BL21-pET21b-tgl在LB培养基中培养至OD6000.5左右,加IPTG诱导,24h后收集菌体并破碎,测定酶活为0.45±0.06U/mL,表明TGase在大肠杆菌中表达后活性得到很大提高。周建[19]、刘凯[20]等人将枯草芽孢杆菌转入大肠杆菌中诱导表达TGase后用Folk比色法法难以测得TGase活性,本实验通过比色法在WB800-pTZ-tgl发酵液中测到了较低的活性,而大肠杆菌表达后活性显著增大,说明所构建的重组菌株TGase得到高效表达。将样品进行SDS-PAGE电泳,结果表明,与对照相比,诱导后BL21-pET21b-tgl在28ku处有一明显表达条带(图9),与TGase推测出的分子量相符,也与相关报道一致[21],证明tgl在BL21中得到了很好的表达。将样品与BSA反应不同时间后进行SDS-PAGE电泳,结果表明随着反应时间延长,BSA发生交联,分子量增大,不但在浓缩胶和分离胶之间形成明显的大分子量蛋白质交联体,而且在点样孔中也发现了明显的蛋白沉积,且远多余WB800-pTZ-tgl发酵液与BSA反应的样品,表明tgl在大肠杆菌中较芽孢杆菌中表达效果更好,且活性明显。与周建[19]等人用重组TGase蛋白催化BSA交联的现像一致,并且本实验证明TGase在70℃下具有高活性,与相关文章表述的该酶具有的高温活性相符[21-22]。
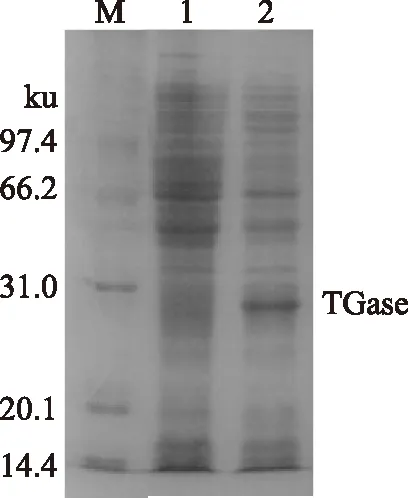
图9 tgl基因在E. coli BL21中的表达Fig.9 Expression of tgl in E. coli BL21注:M:低分子量蛋白Marker;1:未经IPTG 诱导的 BL21-pET21b-tgl;2:经TPTG诱导的BL21-pET21b-tgl。
3 结论与讨论
通过凝胶法初筛及Folk比色法复筛,从土样中分离得到的166株细菌中筛选到一株产谷氨酰胺转氨酶的芽孢杆菌,命名为TGase1318。通过16S rDNA序列比对、生理生化实验和形态学特征分析,鉴定该菌株为枯草芽孢杆菌(B. subtilis)。
用PCR法克隆了该菌株的tgl基因,该基因长738bp,编码的TGase由245个氨基酸组成,分子量为28.29ku,pI 6.71,与来自B.subtilis其他菌株的TGase相比有94%~100%的同源性。
构建了重组质粒pTZ-tgl和pET-21b-tgl,并分别在B.subtilisWB800和E.coliBL21中成功地表达了TGase,WB800-pTZ-tgl发酵液和BL21-pET21b-tgl菌液都能在70℃下催化BSA发生交联反应,大肠杆菌中活性达到0.45±0.06U/mL。
本研究对源于枯草芽孢杆菌的tgl基因成功地进行了异源表达,表达的TGase活性高于已有报道[19-20],并且在70℃下仍表现出明显的对BSA的交联作用,这一特性对高温下的食品加工有潜在的利用价值。但tgl在枯草芽孢杆菌中表达量较低,还有待进一步改善。
[1]Seguro K,Nio N,Motoki M. Some characteristics of a microbial protein cross-linking enzyme:Transglutaminase in macromolecular interaction in food technology[J]. American Chemical Society Symp Ser,1996,650:271-280.
[2]邵虎,陶红军,黄亚东,等.谷氨酰胺转氨酶的研究进展[J].中国酿造,2010(6):9-12.
[3]Lin Y,Chao M,Liu C,et al. Cloning and expression of the transglutaminase gene from Streptoverticillium ladakanum in Streptomyces lividans[J]. Process Biochemistry,2004,39:591-598.
[4]Lin Y,Chao M,Liu C, et al. Cloning of the gene coding for transglutaminase from Streptomyces platensis and its expression in Streptomyces lividans[J]. Process Biochemistry,2006,41:519-524.
[5]Christian K M,Thomas C H,Markus P. Soluble expression of a pro-transglutaminase from Streptomyces mobaraensis in Escherichia coli[J]. Enzyme and Microbial Technology ,2007,40:1543-1550.
[6]kikuchi Y,Date M,Yokoyama K, et al. Secretion of active-form streptoverlicillium mobaraense transglutaminase by Corynebacterium glutamicum:processing of the pro-transglutaminase by a Cosecreted subtilisin-Like protease from Streptomyces albogriseolus[J]. Applied and Environmental Microbiology,2003,69(l):358-366.
[7]Monroe A,Setlow P. Localization of the transglutaminase cross-linking sites in the Bacillus subtilis spore coat protein GerQ[J].Journal of Bacteriology,2006,188(21):7609-16.
[8]de Souza CF,de Matos GS,Flres SH,et al. Environmental effects on transglutaminase production and cell sporulation in submerged cultivation of Bacillus circulans[J]. Applied Biochemistry and Biotechnology,2009,158(2):302-12.
[9]李秀星,戚薇,王建玲.枯草芽孢杆菌谷氨酰胺转胺酶基因的克隆及在大肠杆菌中的表达[J].食品研究与开发,2008,29(6):53-57.
[10]王陆玲,金明晓,宋阳成.产谷氨酰胺转胺酶菌种的筛选鉴定[J].现代食品科技,2010,26(12):1297-1298.
[11]邱秀宝,高东,王颖达.短小芽孢杆菌碱性蛋白酶BP的纯化和性质[J].微生物学报,1994,34(4):293-300.
[12]Anwar A,Saleemuddin M. Alkaline proteases:A review[J]. Bioresource Technology,1998,64:175-183.
[13]东秀珠,蔡妙英.常见细菌系统鉴定手册[M].北京:科学出版社,2001:354-385.
[14]Buehanan R E,N E Gibbons. Bergey’s manual of determinative bacteriology[M]. 8thedition. 1986:The Williams & Wilkins Company.
[15]闫丽娟,赵春雷,谢振荣,等.脂肪酶产生菌Aspergillus niger NJY-1的选育及鉴定[J].生物技术,2009,19(6):17-20.
[16]李瑞芳,薛雯雯,黄亮,等.枯草芽孢杆菌感受态细胞的制备及质粒转化方法研究[J].生物技术通报,2011,5:227-230.
[17]陆雁,王青艳,朱绮霞,等.枯草芽孢杆菌高效转化及其转化子验证方法[J].广西科学院学报,2012,28(2):117-119.
[18]Plácido D,Fernandes C G,Isidro A,et al. Auto-induction and purification of a Bacillus subtilis transglutaminase(Tgl)and its preliminary crystallographic characterization[J]. Protein expression and purification,2008,59(1):1-8.
[19]周建,董亚芳,吴自荣.枯草杆菌谷氨酰胺转胺酶的克隆及其在大肠杆菌中的融合表达[J].中国生物工程杂志,2004,24(8):77-81.
[20]刘凯,刘逸寒,张艳,等.枯草芽孢杆菌谷氨酰胺转氨酶的异源表达[J].天津科技大学学报,2012,27(3):1-5.
[21]Eto Y,Hashiguchi K,Kobayashi K,et al. Bacillus-derived transglutaminase:U.S. Patent 5,731,183[P]. 1998:3-24.
[22]于小娜.产谷氨酰胺转胺酶(TGase)菌株的分离、鉴定及TGase基因的克隆表达[D].南京:南京农业大学,2011:1-76.
Isolation and identification of a transglutaminase producing strain and itstglcloning and expression
ZHANG Ying-ying1,SHI Nan1,2,DU Ping-ping1,SHEN Pei-li1,LI Zhi-hui1,YU Hong-wei1,LU Hai-qiang1,SU Xu-dong1,ZHANG Wei1,TAN Jian-xin1,*
(1.College of Food Science and Technology,Engineering Research Center of Hebei Province for Agricultural Products Processing,Agricultural University of Hebei,Baoding 071001,China;2.College of Life Science,Hebei University,Baoding 071002,China)
By using the dilution plate method,166 bacterial strains isolated from soil samples and one strain,TGase1318,which could produce transglutaminase,was obtained after screening with gel formation method and Folk’s colorimetry method. The strain was identified asBacillussubtilisaccording to the morphology,physiological and biochemical characters as well as 16S rDNA sequence homological analysis. Thetglgene,which was a 738 bp long nucleotide,was cloned from the strain and encoded a 245 residue long TGase. Homological analysis of TGase peptide sequence revealed that it shared 94%~100% conservation with TGase of otherB.subtilisstrains released by NCBI. The expression recombinant plasmids pTZ-tgl and pET21b-tgl were constructed and transformed intoB.subtilisWB800 andE.coliBL21,respectively. The results of BSA crosslink at 70℃ indicated thetglgene was expressed in bothB.subtilisandE.coliwith TGase activity. This study laid a foundation for the application of TGase fromB.subtilisTGase1318 to food industry.
transglutaminase;Bacillussubtilis;cloning and expression;16S rDNA
2014-08-25
张莹莹(1989-),女,硕士研究生,研究方向:环境微生物学。
*通讯作者:檀建新(1968-),男,博士,教授,研究方向:微生物资源开发与利用。
多谷物主食冷冻面团的关键技术研究(13227105D);植物多酚类功能性物质对A(形成的影响(冀人社字[2010]195号)。
TS201.3
A
1002-0306(2015)11-0141-06
10.13386/j.issn1002-0306.2015.11.020
