双毛细管在线干扰校正-电感耦合等离子体发射光谱法测定含铀地质样品中微量铅
2015-05-05贺攀红张延玲孙银生
杨 珍, 贺攀红, 张延玲, 孙银生, 荣 耀, 张 伟
(河南省核工业放射性核素检测中心, 河南 郑州 450002)
双毛细管在线干扰校正-电感耦合等离子体发射光谱法测定含铀地质样品中微量铅
杨 珍, 贺攀红*, 张延玲, 孙银生, 荣 耀, 张 伟
(河南省核工业放射性核素检测中心, 河南 郑州 450002)
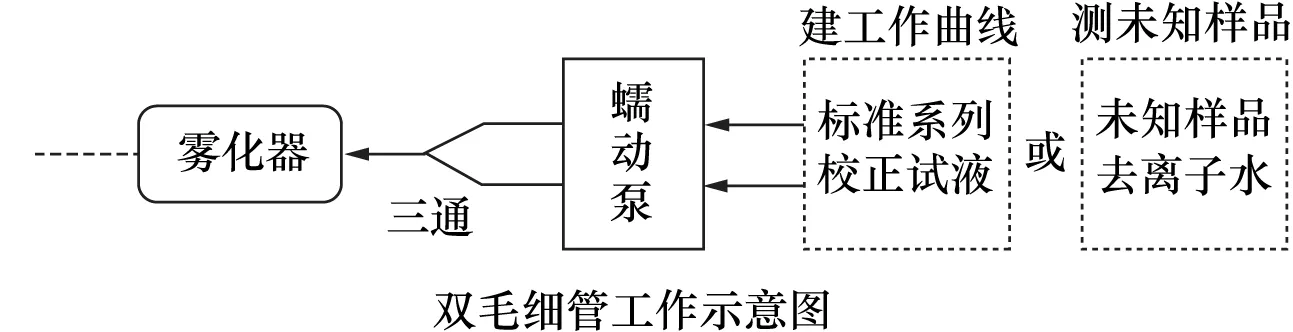
应用电感耦合等离子体发射光谱法测定地质样品中的铅时,基体元素的干扰会使测量结果偏低。本文用氢氟酸、硝酸、高氯酸、盐酸溶解样品,采用等径双毛细管在线干扰校正的方法测定了含铀地质样品中的微量铅。首先通过双毛细管确定了样品溶液中的Fe、Al对铅有负干扰,而一定浓度的U、Ba、Ti、Ca、Mn、K、Mg、Na等基体元素没有干扰或可以忽略,由此在线双毛细管根据样品中Fe、Al的含量使用不同的校正试液建立标准曲线,测定未知样品时同步进行稀释,降低了Fe、Al的基体效应。方法检出限为1.5 μg/g,精密度(RSD)小于5%。与普通干扰校正法相比,双毛细管在线干扰校正法可根据不同基体的样品使用不同的干扰校正试液,快速建立标准曲线进行复杂样品的测定,且避免了二次稀释,节省试剂,适合测定基体成分接近的批量样品。
含铀地质样品; 微量铅; 双毛细管; 在线干扰校正; 电感耦合等离子体发射光谱法
地质样品中铅的测定通常采用电感耦合等离子体发射光谱法(ICP-OES)进行测定[1-4],但是由于存在一定的基体效应致使测定结果偏低。关于基体干扰的消除,罗磊等[5]采用碱熔法分解样品,通过加入氯化钡和优化稀释倍数配合高盐雾化器有效地避免了硫酸钡基体对铅的干扰,方法检出限为0.013%,不能满足低含量铅的检出限要求。陈永欣等[6]采用在标准溶液中加入一定含量Cu、Fe进行基体匹配测定了基体成分相对恒定的铜精矿中的铅,但随着基体类型的变化,需要多次配制标准系列,操作相对繁琐,也存在试剂浪费问题。含铀地质样品中铅的含量一般在0.01%~1%之间,样品经低压密闭酸溶后采用ICP-OES直接测定,铅的测定结果偏低3%~10%,主要是由于Fe、Al等元素造成的基体干扰。
双毛细管采用同时吸取两种不同试液,仅通过改变吸入试液类型即可实现多种测试目的。关于双毛细管的应用,莫桂花等[7]利用提升量比为18的双毛细管在线稀释测定了矿石中的高含量铅,王娟等[8]采用等径双毛细管在线研究了酸度和干扰离子对海带中微量锶测定的影响。本文在确定仪器最佳工作条件的基础上,采用双毛细管在线校正方法查明了样品基体元素对铅的干扰情况,通过在线加入干扰元素的方法建立了标准曲线;在测定样品时将校正试液换为去离子水,对样品同步进行稀释降低了基体效应的影响,实现了含铀地质样品中微量铅的准确、快速测定。
1 实验部分
1.1 仪器及工作条件
iCAP6300电感耦合等离子体发射光谱仪、双毛细管及三通配件(美国ThermoFisher Scientific公司)。仪器工作条件为: 射频功率1150 W,蠕动泵转速50 r/min,辅助气(Ar)流量1.0 L/min,雾化器压力0.18 MPa,水平观测方式,积分时间为15 s。高纯氩气(质量分数大于99.99%)。
铅的分析谱线为220.3 nm。
1.2 标准溶液和主要试剂
铅标准溶液:由1000 μg/mL 铅标准储备液(国家有色金属及电子材料分析测试中心)逐级稀释成标准工作溶液,介质为5%的硝酸。
K、Na、Ca、Mg、Mn、Fe、Al、Ti 、Ba、U工作系列溶液:均由高含量的标准储备液逐级稀释而成,介质为5%的硝酸。
盐酸、硝酸、高氯酸、氢氟酸均为优级纯,实验用水为去离子水(电阻率≥18 MΩ·cm)。
1.3 标准物质和实验样品
本实验涉及的国家一级标准物质有:产铀岩石GBW04118、水系沉积物GBW07311。
实验样品(1#~6#)为核工业系统实验室能力比对样品,由核工业北京地质研究院于2012~2014年间制备;其余测试样品为河南省核工业地质局送检信阳地区含铀地质样品,样品属硅酸盐类。所测样品中各元素的大致含量范围见表1,其中铅的含量范围在几十微克至几千微克之间。
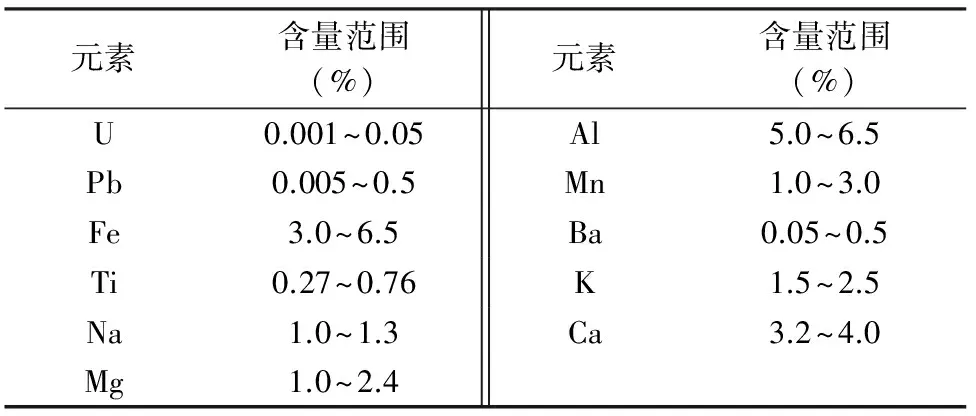
表1 实验样品中的元素含量
1.4 实验方法
准确称取0.1 g(精确至0.0001 g)样品于15 mL聚四氟乙烯管形瓶中,加入氢氟酸4 mL、硝酸2.5 mL、高氯酸0.2 mL,加盖密封在140℃低温电热板上放置48 h后开盖蒸至近干,再加入盐酸1.0 mL,加盖密封2~3 h后开盖蒸至近干,用50%的硝酸5 mL提取1 h,转移至50 mL比色管中,定容,摇匀,待测。同时制备2个样品空白。
采用双毛细管在线加入干扰校正试液建立标准曲线,测定样品时双毛细管一根进样品溶液,另一根进去离子水。
2 结果与讨论
2.1 样品提取酸度试验
在采用四酸低压密闭溶解法中,样品采用硝酸提取,使用国家标准物质GBW07311考察了提取酸度对测定结果的影响。结果表明,在5%~60%硝酸酸度范围内,提取液中铅的测定结果接近,可见酸度影响不大。本法采用50%硝酸5 mL提取后直接定容于50 mL比色管中,使待测溶液保持常用5%的测量酸度。
2.2 光谱干扰
ICP-OES的光谱干扰主要来源于元素谱线之间的重叠[9-10],在选择谱线时尽可能地选择一条干扰少、信号强度高的谱线作为分析谱线。用待测样品溶液做全谱扫描图,通过观察全谱扫描图中分析线的干扰情况,本法选用220.3 nm谱线作为铅的分析线。
2.3 主要基体元素对铅的干扰情况
样品中主要组分Si在四酸全溶矿法过程中已经赶离[11],其他主要基体元素的含量范围在以上表1中已列出。实验考察了含铀地质样品中其他主量元素Fe、Al、K、Na、Ca、Mg、Mn、Ba、Ti以及U对铅的影响。按照地质样品中各元素的大致含量配制0~100 μg/mL的K、Na、Ca、Mg、Mn、Ba、Ti系列溶液,0~300 μg/mL的Fe、Al系列溶液,以及0~1.0 μg/mL的U系列溶液。使用等径双毛细管进行试验,一根进10.0 μg/mL铅标准溶液,另一根进干扰元素系列,在选定分析谱线下进行测定。图1的测定结果显示,样品溶液中含量较高的Al和Fe有负干扰;低于100 μg/mL的Ba、Ti,低于80 μg/mL 的Ca、Mn,低于60 μg/mL的 K、Mg和低于40 μg/mL的 Na的干扰可以忽略;低于1.0 μg/mL的U无干扰。因此干扰校正时主要考虑Fe、Al对铅的影响。
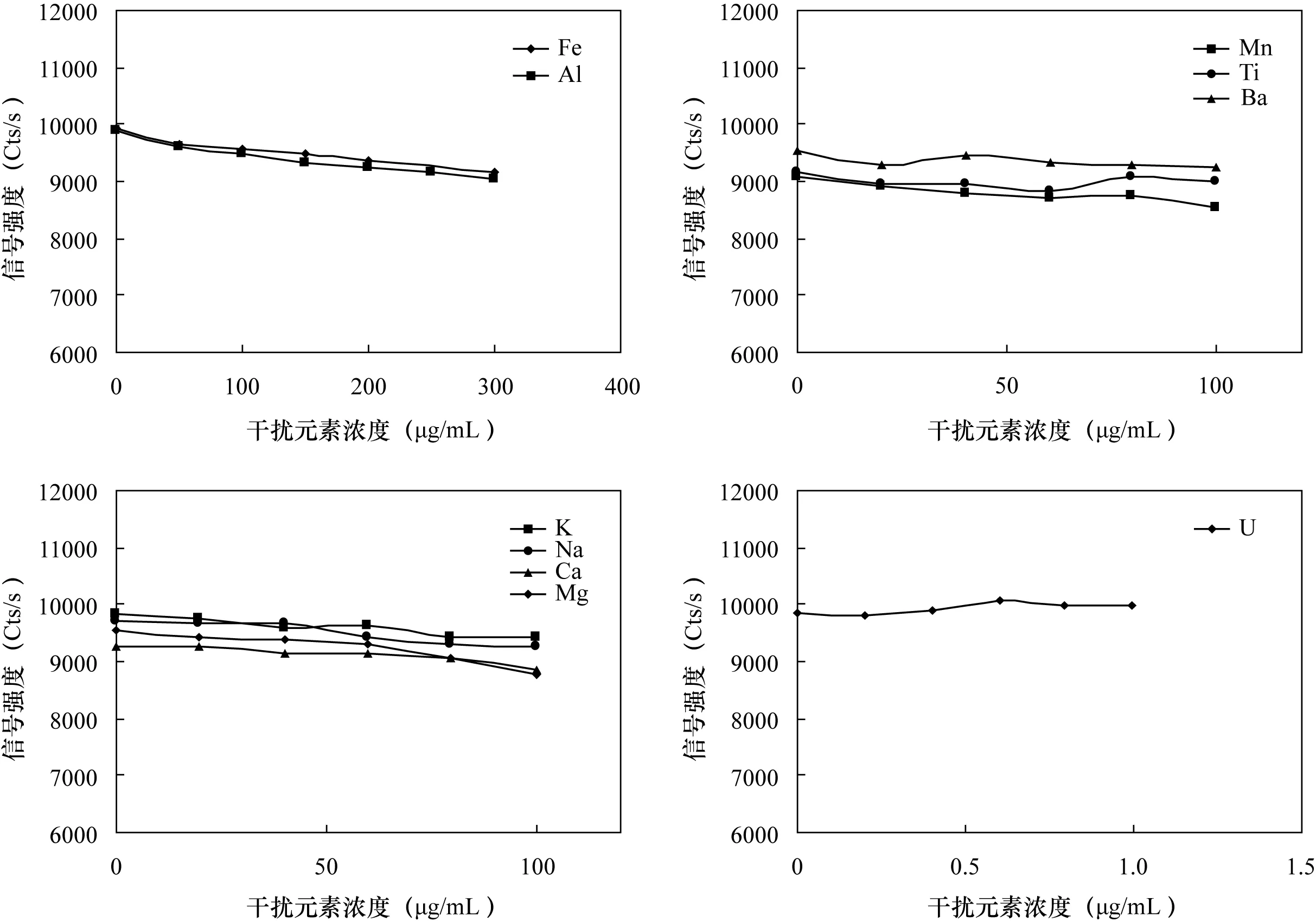
图1 干扰元素的影响Fig.1 Effect of interference elements
2.4 三种干扰校正法的比较
将含铀地质样品溶解后采用直接测定法、普通干扰校正法和双毛细管在线干扰校正法3种方法进行对比实验。直接测定法是用单毛细管吸入纯铅标准溶液建立工作曲线进行样品测定的方法,普通干扰校正法是在铅标准系列溶液中加入等量的混合干扰校正试液,建立工作曲线进行样品测定的方法。而双毛细管在线干扰校正法建立工作曲线时,则是用双毛细管同时吸入纯铅标准系列和混合干扰校正试液,测定样品时将校正试液更换为去离子水。
对于同类型同批次的样品,在样品测量前使用ICP-OES法查明其主量元素Fe、Al的含量[12],根据其含量范围选取折中值配制干扰校正试液,尽可能地使标准溶液和样品溶液中基体元素相匹配。本实验选取1#、2#含铀地质样品,根据这2个样品中Fe、Al的大致含量,配制含Fe浓度为50μg/mL和含Al浓度为100 μg/mL的混合干扰校正试液,按照1.4节实验方法使用双毛细管测定其中铅的含量。
3种校正方法的测定结果(表2)表明:直接测定法测定的铅含量与参考值相比有较大的偏差,经过干扰校正后测量结果与参考值基本吻合。普通干扰校正法和双毛细管在线干扰校正法的结果相差不大。对于双毛细管在线干扰校正法,可根据不同类型样品更换干扰校正试液而不损耗铅标准溶液,更节省标准试剂,同时双毛细管测定有同步稀释的作用,适当倍数稀释对于测定结果有改善[13],本法定容为50 mL,在线双毛细管稀释2倍,与单毛细管吸入相比,降低了样品中基体元素浓度,更好地降低了样品的基体效应。
2.5 方法测定范围和检出限
在100 mL容量瓶中配制0、1.0、10.0 μg/mL铅标准溶液,保持5%硝酸的酸度,在选定的仪器工作条件下采用双毛细管在线加入干扰校正试液建立标准曲线,在0~10.0 μg/mL浓度范围内,铅的标准曲线呈线性,其相关系数为0.9999。
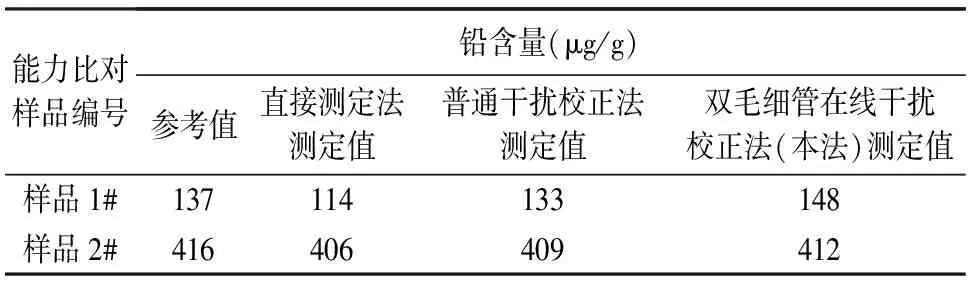
表2 不同干扰校正方法测定铅的结果比较
注:表格中的1#、2#为2012年比对样品,对应列出的“参考值”为中核集团地矿事业部委托核工业北京地质研究院组织的铀矿地质样品分析实验室间比对结果中位值,此中位值是由24家实验室比对结果统计计算得出。
在仪器最佳工作条件下,按1.4节实验方法测定11个试剂空白溶液,计算标准偏差,以3倍标准偏差计算方法的检出限为1.5 μg/g,低于地质分析测试规范DZ/T 0130.4—2006中要求的检出限(2.0 μg/g),与张微等[14]采用酸溶后ICP-OES测定地质样品中Pb相比,检出限略低,表明双毛细管在线校正方法对灵敏度也有所提高。
3 含铀地质样品的分析
按照1.4节实验方法对国家标准物质GBW04118和4个核工业系统实验室间能力比对样品进行测定,测定结果见表3,本法的测定值与参考值基本吻合,相对标准偏差(RSD)小于5%(n=4)。同时分析了3个客户送检样品并进行加标回收实验,加标回收率为98.9%~104.2%(表4)。

表3 国家标准物质和比对样品分析结果
注:表格中的3#、4#、5#、6#为2014年比对样品,对应列出的“参考值”为中核集团地矿事业部委托核工业北京地质研究院组织的铀矿地质样品分析实验室间比对结果中位值,此中位值是由24家实验室比对结果统计计算得出。

表4 实际样品分析结果
4 结论
应用电感耦合等离子体发射光谱法测定地质样品中的微量铅时,含量较多的Fe、Al是主要的干扰元素,其他元素没有干扰或可以忽略不计。双毛细管在线干扰校正法在建立标准曲线时仅通过选择不同浓度的Fe、Al干扰校正试液,即可对不同基体的样品进行测定,对基体成分相近的样品可快速批量测定。与普通的干扰校正法相比,采用在线双毛细管干扰校正法可根据不同基体使用不同的干扰校正试液进行复杂样品的测定,而且样品测定时同步得到稀释,较好地降低了Fe、Al的基体效应,节省了试剂,避免了二次稀释的繁琐操作。
本文所用的在线校正技术、在线稀释技术也适合其他元素测定方法的研究,其不足之处是需要查明样品中基体干扰元素的大致含量,并配制多组干扰校正试液。
[1] 胡德新,王昊云,王兆瑞,等.微波消解样品-电感耦合等离子体原子发射光谱法测定铅精矿中铅、砷、镉、汞[J].理化检验(化学分册),2012,48(7):828-830.
Hu D X,Wang H Y,Wang Z R,et al.ICP-AES Determination of Lead,Arsenic,Cadmium and Mercury in Lead Concentrates with Microwave Assisted Sample Digestion[J].Physical Testing and Chemical Analysis (Part B:Chemical Analysis),2012,48(7):828-830.
[2] 闵国华,张庆建,刘稚,等.电感耦合等离子体原子发射光谱法测定铅矿、锌矿和铅锌矿中杂质元素[J].冶金分析,2014,34(5):51-55.
Min G H,Zhang Q J,Liu Z,et al.Determination of Impurity Elements in Lead Ore,Zinc Ore and Lead-Zinc Ore by Inductively Coupled Plasma Atomic Emission Spectrometry[J].Metallurgical Analysis,2014,34(5):51-55.
[3] 孙晓慧,李章,刘希良.微波消解-电感耦合等离子体原子发射光谱法测定土壤和水系沉积物中15种组分[J].冶金分析,2014,34(11):56-60.
Sun X H,Li Z,Liu X L.Determination of Fifteen Components in Soil and Stream Sediment by Inductively Coupled Plasma Atomic Emission Spectrometry after Microwave Digestion[J].Metallurgical Analysis,2014,34(11):56-60.
[4] 赵刚,谢璐,龙军桥,等.ICP-AES/AFS联合测定金矿地质样品中32种主次痕量元素[J].黄金,2015,36(1):70-74.
Zhao G,Xie L,Long J Q,et al.Combined ICP-AES/AFS Method in Determining 32 Major and Minor Elements in Gold Geological Samples[J].Gold,2015,36(1):70-74.
[5] 罗磊,付胜波,肖洁,等.电感耦合等离子体发射光谱法测定含重晶石的银铅矿中的铅[J].岩矿测试,2014,33(2):203-207.
Luo L,Fu S B,Xiao J,et al.Determination of Lead in Argentalium Ores Containing Barite by Inductively Coupled Plasma-Atomic Emission Spectrometry[J].Rock and Mineral Analysis,2014,33(2):203-207.
[6] 陈永欣,吕泽娥,刘顺琼,等.电感耦合等离子体发射光谱法测定铜精矿中银砷铅锌[J].岩矿测试,2007,26(6):497-499.
Chen Y X,Lü Z E,Liu S Q,et al.Inductively Coupled Plasma-Atomic Emission Spectrometric Determination of Silver,Arsenic,Lead and Zinc in Copper Concentrates[J].Rock and Mineral Analysis,2007,26(6):497-499.
[7] 莫桂花,王志畅,龚治湘.在线双毛细管火焰原子吸收法测定矿石中高含量铅[J].化学工程师,2007(5):26.
Mo G H,Wang Z C,Gong Z X.Determination of High Contents of Pb in Ores by AAS with On-line Bi-capillary[J].Chemical Engineer,2007(5):26.
[8] 王娟,贺攀红,杨珍.在线双毛细管火焰原子吸收法测定海带中的微量锶[J].食品科学,2014,35(10):192-194.
Wang J,He P H,Yang Z.Determination of Trace Strontium in Kelp by Flame Atomic Absorption Spectrometry with On-line Binary Capillary[J].Food Science,2014,35(10):192-194.
[9] 辛仁轩编著.等离子体发射光谱分析[M].北京:化学工业出版社,2005:160-168.
Xin R X.Plasma Emission Spectrum Analysis[M].Beijing:Chemical Industry Press,2005:160-168.
[10] 赵君威,梅坛,鄢国强,等.电感耦合等离子体原子发射光谱分析中的光谱干扰及其校正的研究进展[J].理化检验(化学分册),2013,49(3):364-369.
Zhao J W,Mei T,Yan G Q,et al.Recent Progress of Researches on Spectral Interference and Its Correction in ICP-AES Analysis[J].Physical Testing and Chemical Analysis (Part B:Chemical Analysis),2013,49(3):364-369.
[11] 陈永欣,黎香荣,吕泽娥,等.电感耦合等离子体原子发射光谱法测定锰矿石中铝铜锌铅砷镉[J].冶金分析,2009,29(4):46-49.
Chen Y X,Li X R,Lü Z E,et al.Determination of Aluminium,Copper,Zinc,Lead,Arsenic and Cadmium in Manganese Ores by Inductively Coupled Plasma Atomic Emission Spectrometry[J].Metallurgical Analysis,2009,29(4):46-49.
[12] 邓全道,许光,林冠春,等.微波消解-耐氢氟酸系统进样电感耦合等离子体发射光谱法测定锰矿中铝磷镁铁锌镍[J].冶金分析,2011,31(1):35-39.
Deng Q D,Xu G,Lin G C,et al.Determination of Aluminum,Phosphorus,Magnesium,Iron,Zinc and Nickel in Manganese Ore by Inductively Coupled Plasma Atomic Emission Spectrometry with Hydrofluoric Acid Resistant Sampling System after Microwave Digestion[J].Metallurgical Analysis,2011,31(1):35-39.
[13] 徐进力,邢夏,张勤,等.电感耦合等离子体发射光谱法直接测定铜矿石中银铜铅锌[J].岩矿测试,2010,29(4):377-382.
Xu J L,Xing X,Zhang Q,et al.Direct Determination of Silver,Copper,Lead and Zinc in Copper Ores by Inductively Coupled Plasma-Atomic Emission Spectrometry[J].Rock and Mineral Analysis,2010,29(4):377-382.
[14] 张微,张丽微,艾婧娇,等.ICP-ES法同时测定地质样品中Cu-Pb-Zn-Sc-Mo[J].矿物学报,2013,33(4):521-524.
Zhang W,Zhang L W,Ai J J,et al.Simultaneous Determination of Cu-Pb-Zn-Sc-Mo in the Geological Samples with Inductively Coupled Plasma Optical Emission Spectrometer[J].Acta Mieralogica Sinica,2013,33(4):521-524.
Determination of Trace Lead in Uranium-bearing Geological Samples by Inductively Coupled Plasma-Optical Emission Spectrometry with Double Capillary On-line Interference Correction
YANGZhen,HEPan-hong*,ZHANGYan-ling,SUNYin-sheng,RONGYao,ZHANGWei
(Henan Radionuclide Detection Center of Nuclear Industry, Zhengzhou 450002, China)
Measured lead contents in geological samples were commonly lower than real values because of matrix elements interference during Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) analyses. In this study, U-bearing geological samples were dissolved by hydrofluoric acid, nitric acid, perchloric acid, and hydrochloric acid. Trace lead was determined by ICP-OES with double capillary on-line interference correction. The interferences of matrix elements on lead were studied using double capillary. Fe and Al had negative interferences on lead, whereas a specific concentration of U, Ba, Ti, Ca, Mn, Mg, K and Na almost had no interferences on lead. The standard curve was established using double capillary according to the contents of Fe and Al in different test solution. The unknown sample was diluted simultaneously, which reduced the matrix effects of Fe and Al. The detection limit was 1.5 μg/g, and the RSD was less than 5%. Compared with the common interference correction method, the double capillary on-line interference correction method can establish a standard curve quickly using different interference correction test solutions according to different matrices. Moreover, sample solution does not need to be secondly diluted, saving the reagent. This method is suitable for rapid determination of batch samples with similar matrices.
geological samples with uranium; trace lead; double capillary; on-line interference correction; Inductively Coupled Plasma-Optimal Emission Spectrometry
2015-05-08;
2015-08-20; 接受日期: 2015-09-05
杨珍,工程师,主要从事岩矿分析工作。E-mail: 24291232@qq.com。
贺攀红,工程师,主要从事岩矿分析工作。E-mail: hepanhong1983@163.com。
0254-5357(2015)05-0528-05
10.15898/j.cnki.11-2131/td.2015.05.005
O614.433; O657.31
A
