埃罗替尼盐酸盐的全合成
2015-04-23吴海龙朱思兰蔡晓阳史海健
吴海龙,朱思兰,蔡晓阳,陈 程,朱 晶,史海健
(南京工业大学化学化工学院,江苏南京 210009)
埃罗替尼(7)是以表皮生长因子受体为作用靶点的抗癌药物[1-3],在临床实践和研究中具有广泛应用[4-6]。
7的结构主要由喹唑啉母体骨架、芳胺取代基和侧链烷氧基三部分组成,为了便于保存通常也将其制备成埃罗替尼盐酸盐(7·HCl)。根据不同的策略,7的合成方法主要有芳胺亲核取代法[7-10]和汇聚法[11-12]。
通过对7·HCl的逆合成分析,本文设计了一条新的合成7·HCl的路线。即以3,4-二羟基苯甲酸(1)为起始原料,首先用2-氯乙基甲基醚将侧链醚化得3,4-二(2-甲氧基乙氧基)苯甲酸(2);2中的羧基先与氯化亚砜反应,再与浓氨水作用转化为酰胺基得3,4-二(2-甲氧基乙氧基)苯甲酰胺(3);3在五氧化二磷的作用下脱水得3,4-二(2-甲氧基乙氧基)苯甲腈(4);4用硝酸硝化引入硝基得4,5-二(2-甲氧基乙氧基)-2-硝基苯甲腈(5);5中的硝基用连二亚硫酸钠还原为氨基则得到汇聚成环反应底物2-氨基-4,5-二(2-甲氧基乙氧基)苯甲腈(6);6先与DMF-DMA作用,再与3-乙炔基苯胺反应一步完成喹唑啉骨架的构建和芳胺取代基的引入得7;7与HCl成盐合成了目标化合物7·HCl(Scheme 1),其结构经1H NMR,IR和MS确证。
1 实验部分
1.1 仪器与试剂
X-4型显微熔点仪(温度未校正);Bruker MD 300 MHz和500 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Nicolet AVATAR-360型红外光谱仪(KBr压片);VG-ZAB-MS型质谱仪(70 eV)。
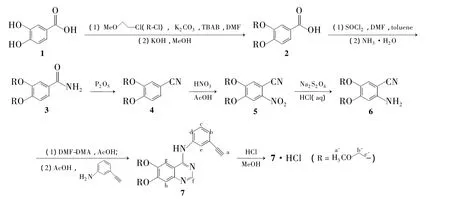
Scheme 1
所用试剂均为分析纯。
1.2 合成
(1)2的合成
在三口烧瓶中加入 1 10.0 g(65 mmol),K2CO335.9 g(0.26 mol),四丁基溴化铵(TBAB)0.72 g(2.23 mmol)及 DMF 120 mL,搅拌下于100℃反应1 h。降温至50℃,滴加2-氯乙基甲基醚23.6 mL(0.26 mol),滴毕,于 95 ℃ 反应 12 h(TLC监测)。冷却至室温,过滤,滤饼用DMF洗涤,合并滤液与洗液,减压蒸除溶剂得棕褐色油状物。用混合溶剂[KOH(10.9 g)+MeOH(105 mL)+H2O(35 mL)]溶解,于室温反应过夜。减压蒸除甲醇,于0℃用2 mol·L-1盐酸调至pH 3。析出固体,过滤,滤饼用蒸馏水洗涤,于室温真空干燥得白色固体 2 12.36 g,收率 70%;1H NMR δ:3.46(s,6H),3.79 ~3.81(m,4H),4.20 ~ 4.24(m,4H),6.93 ~6.94(d,J=5.0 Hz,1H),7.63 ~ 7.65(d,J=10.0 Hz,1H),7.72 ~7.73(d,J=5.0 Hz,1H);IR ν:2 975,2 919,2 813,1 665,1 598,1 447,1 277,1 233,870,762 cm-1。
(2)3的合成
在三口烧瓶中加入2 12.36 g(46 mmol),甲苯120 mL和 DMF 0.75 mL,搅拌下加入 SOCl233.4 mL(0.46 mol),回流反应 10 h。降至室温,滴入冰冷浓氨水中,立刻有白色固体形成。过滤,滤饼用水洗涤,于室温真空干燥得白色固体3 7.5 g,收率 61%;1H NMR δ:3.44(s,6H),3.78(m,4H),4.20(m,4H),5.85(brs,2H),6.90 ~6.91(d,J=5.0 Hz,1H),7.32 ~ 7.34(d,J=10.0 Hz,1H),7.46(s,1H);IR ν:3 367,3 175,2 990,2 934,2 887,2 822,1 648,1 439,1 265,1 121,870,800 cm-1。
(3)4的合成
在三口烧瓶中加入3 3.0 g(11 mmol),二甲苯40 mL及P2O54.7 g(33 mmol),搅拌下回流反应30 min(TLC监测)。降温至80℃,趁热过滤,滤饼用热乙酸乙酯(50 mL)洗涤,合并滤液与洗液,用饱和Na2CO3溶液(2×10 mL)洗涤,无水Na2SO4干燥;减压蒸除溶剂得淡黄色油状液体4 1.86 g,收率 66%;1H NMR δ:3.45(s,6H),3.75 ~ 3.81(m,4H),4.14 ~ 4.22(m,4H),6.92 ~6.94(d,J=10.0 Hz,1H),7.14 ~7.16(d,J=10.0 Hz,1H),7.24 ~7.27(d,J=15.0 Hz,1H);IR ν:2 937,2 884,2 816,2 225,1 515,1 450,1 271,1 121,1 044,806 cm-1。
(4)5的合成
在反应瓶中加入浓硝酸4.1 mL,搅拌下于0℃滴加4 1.86 g(7.40 mmol)的乙酸(3.5 mL)溶液,滴毕,于50℃反应3.5 h(TLC监测)。倒入冰水(20 mL)中,有淡黄色固体析出;过滤,滤饼依次用冰水(20 mL),正己烷(20 mL)洗涤,于室温真空干燥得淡黄色固体5 1.84 g,收率84%;1H NMR δ:3.45(s,6H),3.82(s,4H),4.29 ~4.30(m,4H),7.26(s,1H),7.86(s,1H);IR ν:2 928,2 887,2 222,1 527,1 336,1 297,1 218,1 050,894 cm-1;MS m/z:319{[M+Na]+,100%}。
(5)6的合成
在三口烧瓶中加入 5 1.0 g(3.38 mmol),Na2S2O43.53 g(20.28 mmol)和水 12 mL,搅拌下于50℃反应4 h(TLC监测)。滴加浓盐酸6.5 mL,滴毕,用50%NaOH溶液调至pH 10。用乙酸乙酯(2×25 mL)萃取,合并萃取液,用饱和食盐水洗涤,无水硫酸钠干燥;减压蒸除溶剂得棕色固体 6 0.8 g,收率 89%;1H NMR δ:3.43(s,6H),3.70 ~ 3.76(m,4H),4.05 ~ 4.12(m,4H),6.28(s,1H),6.91(s,1H);IR ν:3 455,3 358,2 890,2 816,2 198,1 518,1 433,1 268,1 127,865 cm-1。
(6)7的合成
在三口烧瓶中加入 6 0.8 g(3.0 mmol),甲苯15 mL,乙酸10 mL,DMF-DMA(N,N-二甲基甲酰胺二甲缩醛)0.8 mL(6.0 mmol),搅拌下于105℃反应1.5 h(TLC监测)。减压蒸除甲苯和DMF-DMA,依次加入甲苯15 mL,3-乙炔基苯胺0.33 mL(3.0 mmol)和乙酸0.35 mL(6.0 mmol),回流反应10 h(TLC监测)。倒入冰水中,用氨水调至pH 8,有褐色固体析出;过滤,滤饼依次用冰水(20 mL)和正己烷(20 mL)洗涤,用乙酸乙酯重结晶得淡棕色固体7 0.86 g,收率68%;1H NMR δ:3.09(s,1H),3.45(s,6H),3.82(s,4H),4.23 ~ 4.30(m,4H),7.21 ~ 7.34(m,3H),7.65(brs,1H),7.76 ~ 7.78(d,J=10.0 Hz,1H),7.89(s,1H),8.62(s,1H);IR ν:3 275,2 925,2 884,1 506,1 427,1 241,1 203,1 121,859,785 cm-1;MS m/z:394{[M+H]+,100%}。
(7)7·HCl的合成
在反应瓶中依次加入7 0.8 g(2.03 mmol)和甲醇15 mL,搅拌加热使其溶解;于25℃通入干燥HCl气体0.5 h,有淡黄色固体析出;过滤,滤饼用甲醇20 mL洗涤,于室温真空干燥得淡黄色固体 7·HCl 0.8 g,收率 92%,纯度 99.1%(HPLC);1H NMR(DMSO-d6)δ:3.36(s,6H,a'-H),3.78 ~3.80(t,J=10.0 Hz,4H,b'-H),4.26(s,1H,a-H),4.32 ~4.34(t,J=10.0 Hz,2H,c'-H),4.38 ~4.40(t,J=10.0 Hz,2H,c'-H),7.40(s,1H,b-H),7.42(s,1H,e-H),7.48 ~7.51(t,J=7.5 Hz,1H,c-H),7.78 ~7.80(d,J=10.0 Hz,1H,d-H),7.88(s,1H,h-H),8.40(s,1H,g-H),8.84(s,1H,f-H),11.44(s,1H,HCl);IR ν:3 269,3 008,2 904,2 825,2 695,2 645,1 633,1 571,1 509,1 444,1 274,1 121,870 cm-1;MS m/z:394{[M -Cl]+,100%}。
2 结果与讨论
2.1 合成
(1)2的合成
1与2-氯乙基甲基醚的Williamson反应是固液非均相反应,故需用相转移催化剂TBAB;由于两个酚羟基在苯环上互为邻位,第一个羟基醚化相对比较容易,而第二个羟基的醚化由于空间位阻的存在而变得困难,因此进行了反应条件的优化。实验结果表明:合成2的最佳反应条件为:1 65 mmol,n(1)∶n(2-氯乙基甲基醚)=1 ∶4,于95℃反应12 h,收率70%。在此条件下合成的2,在碱性条件下水解完全后用2 mol·L-1盐酸酸化,纯度很高,可直接用于下步反应。
(2)3的合成
3的合成分两步:2先与氯化亚砜反应生成其酰氯(A);A与氨反应生成 3。2 1.9 mmol,n(2)∶n(SOCl2)=1 ∶10,回流反应 10 h,考察溶剂对反应的影响,结果见表1。由表1可知,相同的反应时间下,2在三种不同溶剂中的转化率差别很大,在甲苯中转化率最高(95%)。甲苯为酰氯化反应的最佳溶剂。

表1 溶剂对3收率的影响*Table 1 Effect of solvent on yield of 3
以甲苯为溶剂,其余反应条件同2.1(2),考察SOCl2用量[r=n(2)∶n(SOCl2)]对反应的影响,结果见表2。由表2可见,当r=1∶6和1∶8时,2的转化率还有上升的空间;当r=1∶10时,转化率达95%;继续增加氯化亚砜用量,转化率不再上升。因此,从成本以及后处理的难度考虑,最佳的r=1∶10,两步反应收率为61%。
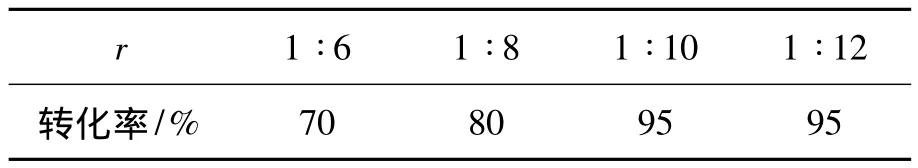
表2 r对3收率的影响*Table 2 Effect of r on yield of 3
(3)5~7的合成
5的合成采用65%浓硝酸的醋酸溶液作为硝化试剂。反应条件优化结果为:当 n(4)∶n(HNO3)=1∶8时,5的收率达63%。5用连二亚硫酸钠还原,在最佳投料比n(5)∶n(Na2S2O4)=1∶6时,6的收率达89%。
7的合成是通过会聚反应构建喹唑啉骨架并引入3-乙炔基苯胺一锅内完成,是整条合成路线中最为关键的步骤。在甲苯和催化量的乙酸中,7用DMF-DMA处理得到甲脒中间体,将多余的DMF-DMA和溶剂甲苯经减压蒸馏除去,再于同一锅内加入3-乙炔基苯胺,过量的乙酸和溶剂甲苯,接着回流反应10 h。冰水中并用氨水调节pH至7,棕色固体析出,再用甲醇重结晶,收率68%。
3 结论
本文采用较为新颖的会聚法合成策略。首先将侧链醚化,然后经四步反应得到关环底物6;6在关键的会聚成环反应中一锅法合成7;7最后与HCl气体成盐得到7·HCl。
该合成路线设计思路简洁巧妙,基本符合预期的设计目的,经过工艺优化后可适用于大规模工业生产,具有潜在的市场价值。
[1]Pawar V G,Sos M L,Rode H B,et al.Synthesis and biological evaluation of 4-anilinoquinolines as potent inhibitors of Epidermal growth factor receptor[J].J Med Chem,2010,(53):2892 -2901.
[2]Grandis J R,Sok J C.Signaling through the Epidermal growth factor receptor during the development of malignancy[J].Pharmacol Ther,2004,(102):37 -46.
[3]Krause D S,Van Etten R A.Tyrosine kinases as targets for cancer therapy[J].N Eng J Med,2005,(353):172-187.
[4]Brahimi F,Matheson S L,Dudouit F,et al.Inhibitor of Epidermal growth factor receptor-mediated signaling by“Combi-triazene”BJ2000,a new prode for combitargeting postulates[J].J Phar Exp Ther,2002,(303):238-246.
[5]Festuccia C,Gravina G L,Biordi L,et al.Effects of EGFR tyrosine kinase inhibitor Erlotinib in prostate cancer cells in vitro[J].The Prostate,2009,(69):1529-1537.
[6]Yamasaki F,Zhang D W,Bartholomeusz C,et al.Sensitivity of breast cancer cells to Erlotinib depends on cyclin-dependent kinase 2 activity[J].2007,(6):2168-2177.
[7]Knesl P,Roseling D,Jordis U.Improved synthesis of substituted 6,7-dihydroxy-4-quinazolineamines:Tandutinib,Erlotinib and Gefitinib[J].Molecules,2006,11(4):286-297.
[8]Marzaro G,Guiotto A,Pastorini G,et al.A novel approach to quinazolin-4(3H)-one via quinazoline oxidation:An improved synthesis of 4-anilinoquinazolines[J].Tetrahedron,2010,66(4):962 -968.
[9]Chandregowda V,Rao G V,Reddy G C.One-pot conversion of 2-nitrobenzonitriles to quinazolin-4(3H)-ones and synthesis of Gefitinib and Erlotinib hydrochloride[J].Heterocycles,2007,71(1):39 -48.
[10]Chandregowda V,Rao G V,Reddy G C.Improved synthesis of Gefitinib and Erlotinib hydrochloride-anticancer agents[J].Synthetic Communications,2007,(37):3409-3415.
[11]Chandregowda V,Rao G V,Reddy G C.Convergent approach for commercial synthesis of Gefitinib and Erlotinib[J].Organic Process Research & Developent,2007,(11):813 -816.
[12]Asgari D,Aghanejad A,Mojarrad J S.An improved convergent approach for synthesis of Erlotinib,a tyrosine kinase inhibitor,via a ring closure reaction of phenyl benzamidine intermediate[J].Bull Korean Chem Soc,2011,(32):909 -914.
