糖尿病性心肌病的发病机制和治疗方法
2015-04-06王贵法
王贵法
作者单位: 050011河北省石家庄市第一医院重症医学科
糖尿病性心肌病的发病机制和治疗方法
王贵法
作者单位: 050011河北省石家庄市第一医院重症医学科
【关键词】糖尿病性心肌病;高血糖;脂肪酸;蛋白激酶C;醛固酮;自主神经病
由于肥胖和久坐的生活方式,预计到2025年将会有3亿人罹患糖尿病。糖尿病患者的死亡80%是由于心血管疾病,而这其中大部分是由冠心病引起的。然而,现在有一种看法越来越得到认可,即糖尿病患者还遭受另外一种心脏损害,称为“糖尿病性心肌病”。这一概念最初是在1972年提出的,它主要是基于对四位有心衰却不伴高血压,冠心病,心瓣膜病,先天性心脏病的糖尿病患者的观察,对这一疾病的承认也获得了很多流行病学和分子方面的资料,及大量诊断研究的支持[1]。
1 流行病学
Karamitsos等[2]证实充血性心力衰竭发病率升高,在男性糖尿病患者为2.4∶1,在女性糖尿病患者为5∶1,这一结果与年龄,高血压,肥胖,CAD及高脂血症无关。其他前瞻性研究也表明了糖尿病患者发生心力衰竭的终生危险增加,以及死于Q波和无Q波心肌梗死的危险也升高.这些均表明,糖尿病心肌受到另外一种损害,它造成了心肌更大的伤害,并继发心力衰竭。在社区人群中,糖尿病患病率是4%~6%,与之不同的是,在参加SOLVD(studies of left ventricular dysfunction; 26%),ATLAS (assessment trial of lisinopril and survival; 19%)and V-HeFTⅡ(vasodilator-heart failure trialⅡ; 20%)等心衰试验的人群中糖尿病患者的比例却远远超出,从而也证明了糖尿患者群心力衰竭发病危险的增加[3]。
2 病理学
在伴有糖尿病的患者中,致死性和非致死性冠心病的发生率增加了2~4倍,尸检发现糖尿病患者比不伴糖尿病的冠心病患者,其冠心病更广泛。血管造影也发现有更多严重的近中和远侧的冠心病。伴有糖尿病的患者冠心病病死率增加3~7倍,有80%的糖尿患者人死于冠心病。在糖尿患者中,心梗后近期和远期病死率增加了1.5~2倍。而且,无心梗病史的2型糖尿病患者,与曾发过心梗的非糖尿病患者的心梗发生危险同样高。同样的,因不稳定心绞痛住院的患者登记结果显示,无心血管疾病史的糖尿病患者与不伴糖尿病的CAD患者的远期发病率和病死率相同[3]。而且,与不伴糖尿病的患者相比,尽管梗死面积相近,但糖尿病患者梗死后心衰的发病率更高。梗死后,非糖尿病患者的幸存心肌过度代偿,以维持心脏输出。然而在糖尿病患者中,幸存的心肌并不能完成这一补偿功能,因为已经有复杂的内源和外源性因素降低了冠状动脉血流储备功能。此外,从糖尿病患者的心内膜心肌取样显示有毛细血管基底膜增稠,心肌细胞萎缩及肥大,并伴有心肌及间质纤维化,进一步降低心肌功能。
3 定义
糖尿病患者心肌发生大范围结构改变最后引起左心室肥大,心脏舒张和收缩功能障碍。这一疾病过程称为糖尿病性心肌病。糖尿病使心肌在细胞水平上发生改变,从而导致结构异常,糖尿病性心肌病这一概念即是基于此。我们知道,糖尿病患者发生高血压和冠心病的危险增加。糖尿病性心肌病观点认为这些改变会发生,在不伴有其他因素的情况下可以被检测到。因此,高血压和冠心病患者会因为以上的疾病过程发生心肌改变,但是一种特异性心肌病也会继发于糖尿病去影响心肌,从而造成糖尿病和高血压联合的有害作用。糖尿病性心肌病是亚临床还是临床取决于它的症状和体征。在症状出现前,它将经过一个很长的亚临床过程[4]。
4 糖尿病性心肌病的分子基础
高血糖,高脂血症和活性氧(ROS)增多诱发下游区转录因子改变,导致基因表达,心肌底物利用,心肌生长,内皮功能,心肌顺应性等改变(图1)[5]。
4.1高血糖高血糖可能以一连串次级转换反应介导其损伤效应。一个基本异常便是糖基化终末产物(AGEs)的过度生成,AGEs能钝化NO,损伤冠状血管扩张。持续高血糖致使线粒体ROS生成过多,从而影响转录,导致收缩期功能障碍。ROS过度减少NO水平,会导致心肌炎症和内皮功能障碍,这一过程是通过PARP[poly (ADP-ribose)polymerase])进行的,而抑制PARP可以逆转糖尿病内皮功能障碍[6]。
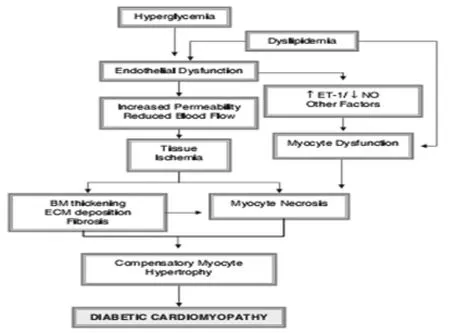
舒张期功能障碍的严重程度与糖化血红蛋白(HbA1c)水平相关,可能的原因是AGE诱导ROS生成,使心肌胶原沉着和纤维变性。金属硫蛋白表达增加,它是一种有效的抗氧化剂,能限制糖尿病性心肌病的发展,阻止胶原交联,改善舒张功能。最近,糖基化过程被认为直接与心肌钙管理及收缩性有关.SERCA2a(sarcoplasmic/endoplasmic-reticulum Ca2 +-ATPase 2a)负责细胞内钙释放后的补充,它使心肌收缩停止,故它是心脏舒张所必需的。SERCA2a是一种P-type ATPase,它利用ATP末端磷酸键水解释放的能量,逆电化学梯度将钙泵入。SERCA2a更新率低,所以它对翻译后修饰很敏感,特别是在糖尿病这样的慢性病程中。因此SERCA2a的糖基化导致其活性下降,使心脏舒张期延长[7]。
4.2脂肪酸不依赖于高脂血症对冠状动脉内皮功能的影响,糖尿病心肌中脂肪酸供给增加导致了数个主要的细胞代谢紊乱。β氧化增加和线粒体长链脂酰肉毒碱蓄积,致使氧化磷酸化解耦联。脂肪酸氧化增强可通过抑制丙酮酸脱氢酶,减少葡萄糖和丙酮酸利用。丙酮酸氧化可被pdk4(冬眠基因)进一步减少,被PPAR (peroxisome-proliferator-activated receptor)激活。净结果是糖酵解的中间产物过多,神经酰胺合成增加诱导凋亡,PPAR-α和-γ促效药曲格列酮阻止[8]。糖酵解,丙酮酸氧化,乳酸摄取,以及作为乙酰辅酶A来源,这一些作用受损导致了心肌生物能学和舒张/收缩耦联的紊乱。传统的药物治疗主要是针对恢复ATP合成和氧输送之间的平衡(长效硝酸盐或钙离子拮抗剂),或是通过降低血压和心率来减少心脏做功(钙离子拮抗剂或B-受体阻滞剂)。
4.3蛋白激酶(PKC)二脂酰甘油(DAG)过度激活活化了PKC信号转导途径,这一过程已在糖尿病动物血管组织中识明,使血管细胞对高糖易感[9]。它会引起糖尿病性心肌病的众多改变,包括组织血流量减少,增加细胞外基质沉着,毛细血管基底膜增厚,血管渗透性增加伴新生血管形成。LY333531,一种PKCβ特异性抑制剂,在许多Ⅱ期临床试验中已被证实是对糖尿病视网膜病,肾病,神经病变以及心脏功能障碍均有效果[3]。
4.4RAS刺激心脏牵张感受器使肾素血管紧张素系统(RAS)和交感神经系统(SNS)激活,导致心肌机构和构型改变,损害了心脏做功能力。在糖尿病中,尽管有心脏负荷的轻度改变,RAS却有增量调节。在糖尿病鼠中,血管紧张素Ⅱ表达过多导致了心肌肥大和细胞凋亡。促生长因子-1(IGF-1)能够下调p53基因的表达,减少血管紧张素原的转录,从而降低血管紧张素Ⅱ,使心肌细胞凋亡减少。同样地IGF-1的过表达也能下调RAS系统,而抑制糖尿病性心肌病的发展[10]。CEI在预防和逆转糖尿病性心肌病的效果在增加,这些发现给其作用提供了分子基础.
4.5醛固酮致纤维化小型糖尿病临床研究已证明,用醛固酮拮抗剂治疗心衰能降低心血管病死率。在RALES (Randomized Aldactone Evaluation Study)试验中,充血性心衰患者在其标准治疗(ACEI,袢利尿剂,地高辛等)中加入安体舒通,可以降低30%的病死率[11]。这一效果只在血清胶原合成标记水平升高的患者中比较明显,它提示了限制细胞外基质过多转归可能是安体舒通起效的一方面机制。已知血管紧张素Ⅱ和醛固酮能致使心脏纤维化。在活体内,它是以胶原堆积增多和纤维母细胞增殖增加为特征的。在体外,血管紧张素Ⅱ和醛固酮通过人成心肌细胞剂量依赖性地增加胶原合成。另外,血管紧张素Ⅱ也能刺激人成心肌细胞增殖。局部肾素-血管紧张素-醛固酮系统存在于了所有成分,也包括了酶和受体,这一点已被证实。另外,在2个月老鼠和人类胎儿的心脏中也有醛固酮存在。糖尿病患者心脏微血管病变致使心肌广泛纤维化没,这一改变成为糖尿病性心肌病。通过糖尿患者死后对其心脏组织检验,发现其间质和局灶性血管周围均有胶原堆积,表现出纤维化。在肾素-血管紧张素-醛固酮系统失调特别是伴高血糖患者中,醛固酮和葡萄糖通过刺激肌纤维母细胞生长来调节纤维化。
4.6HIF-1(hypoxia-inducible factor-1)/VEGF(vascular endothelial growth factor)糖尿病患者心肌对缺血的一个不利的血管性反应,导致侧支形成差,故易于梗塞。正常情况下,血管细胞是静止的,但可被不同刺激物所激活,尤其是缺氧(发生于局部缺血或梗死中)。缺氧主要是由HIF-1来介导的,HIF-1是一种转录调节复合物,它通过一个特异的启动序列[HRE(hypoxia response element)]来起作用,这一序列存在于很多基因启动子,包括VEGF。通常被蛋白酶降解,缺氧和自由基稳定HIF-1α,并使它活化[12]。它控制多种血管原性生长因子的表达,包括血管生成素-1,-2,-4,PGF (placental growth factor ),PDGF-β(platelet-derived growth factor-β)and VEGF。一些观察认为VEGF在心脏损伤应答中起着重要的作用。心梗后,VEGF mRNA在心肌细胞,血管平滑肌细胞和浸润巨噬细胞中显著性升高。在急性缺血或早期梗死的患者中,HIF-1α mRNA被增量调节,在随后的明确梗死阶段可见到VEGF转录体[13]。不同地是在糖尿病中,糖尿病和非糖尿病胰抗老鼠的心肌中VEGF mRNA和蛋白及其受体VEGF-R1,VEGF-R2的表达有明显下降(40%~70%);与非糖尿病患者相比,糖尿病患者心室中VEGF-R1,VEGF-R2下降2倍。提示糖尿患者正常的血管发生的分子过程受损[3]。
4.7基因表达在糖尿病性心肌病中,许多关键分子发生基因改变。在链脲佐菌素(STZ)糖尿病鼠改善高血糖6周后发现,心肌中肉毒碱棕榈酰基转移酶1~8及另外新的序列的基因表达增强,预计将会在信号转导中起作用。糖尿病动物心肌的氧化损伤归因于WAF1/CIP1基因编码的一种蛋白,它能抑制大量cyclin-CDK复合物[14]。糖尿病性心肌病的发展过程中,肌纤维膜的Na+/K+ATPase,Na+/Ca2 +交换,及Ca2 +泵活性的异常会导致细胞内钙超负荷。alloxan诱导的糖尿病鼠的心室肌中可观察到,Na+/K+ATPase α1亚单位mRNA明显减少,Na+/Ca2 +交换剂mRNA增加。糖尿病性心肌病的一个关键的电生理异常是致心律失常性升高,它可能与复极K+电流减少有关。
4.8内皮功能障碍内皮功能障碍是动脉粥样硬化的前身和结果。糖尿病通常有血管内皮解剖和功能上的异常。慢性高血糖和血脂障碍会促成内皮功能障碍。高血糖导致内皮细胞NO生成受损,前列腺素,糖基化蛋白,内皮黏附分子,血小板和血管生长因子生成增加,它们累积地增强了血管紧张度,增加血管通透性以及促其生长和构型重建。内皮功能障碍也包括毛细血管内皮加速消失,削弱了细胞间联系,改变了蛋白质合成,改变了内皮细胞附着糖蛋白合成与表达,它能够促进单核细胞和白细胞的附着和它们的跨内皮移行。此外高血糖增加了内皮细胞基质产生,这可能与基底膜增厚有关。内皮功能障碍的临床表现不仅仅是动脉粥样硬化增加.内皮细胞也有助于侧支循环的形成,这一功能在糖尿病中是被降低的,这可能会解释在这些糖尿病患者中梗死范围增加和心梗后充血性心衰发病增加的现象。
4.9动脉僵度高血压和糖尿病通过内皮功能障碍介导的纤维化导致动脉僵度增加。Vinereanu等[15]证实在动脉僵度和受损的左心室功能间存在联系。他们的研究结果指出,在伴有动脉僵硬,相对无顺应性的患者中,左心室心内膜下功能可能被抑制。大动脉顺应性降低影响了波反射的时限,影响了心室负荷。心室射血产生了一个向前的压力波,它可以被动脉树反射回来。在动脉僵度增高的患者中,这道反射波返回过早,还处于左心室射血期,从而使中心收缩压和左心室后负荷增加,导致中心舒张压和冠状动脉灌注压降低。这些血液动力学改变的净效应就是局部缺血.
4.10自主神经病变(CAN)可能会使舒张功能受损,并使糖尿病患者心血管危险增加。糖尿病自主神经病变会导致冠状阻力血管对交感神经刺激增加的血管舒张反应受损。21%的不伴缺血性心脏病的1型糖尿病患者有舒张期充盈异常,且与CAN严重程度相关[16]。同样地,在一些自主神经患者中突出表现为心室充盈异常。2型糖尿病中交感神经功能障碍与舒张和收缩功能障碍均有关。站立后异常的收缩期血压与减少的二尖瓣E/A ratio有关。研究发现糖尿病患者中,深呼吸时平均心率变异显著减少和舒张期最大充盈率异常关联,也反映了副交感神经和心脏功能障碍存在联系。自主神经病患者二尖瓣E/A ratio明显降低,E/A ratio和自主神经病显著相关[16]。
5 治疗
5.1血糖控制血糖控制差将会导致心血管病死率危险增加,HbA1c水平每升高1%,心血管病死率增加11%[17]。最近一项研究证实HbA1和HF相关。改善血糖控制对心血管发病率和病死率将其起到积极效果。但UKPDS未能得出,使用磺脲类或胰岛素强化血糖控制对减少2型糖尿病患者发生大血管病危险性没有显著益处。应该看到UKPDS在设计方法有显著的局限[17]。这一研究是非盲法的,而且当在最初议定时限内分析无区别时,研究继续进行。仅接受饮食治疗组的患者实际上在空腹血糖>15 mmol/L时接受药物治疗,在9年里只有25%患者接受单一治疗。然而在二甲双胍治疗超重(>120%理想体重)患者小组却获得了显著效果,糖尿病相关病死率(42%),任何原因病死率(36%)心肌梗死(39%)。但这些数据应该谨慎对待,因为它们来自高度选择的分组(有明显心血管病的患者除外)。然而这些数据能够平息一些来自NIH-funded UGDP研究的担心,这一研究报道了磺脲类导致心血管病死率增加。磺脲类作用机制是关闭B细胞上钾离子通道以增加胰岛素释放,然而在心肌细胞和血管平滑肌细胞上它们也起着同样作用,而至少在理论上可能会对局部缺血的预处理和血管舒张有不利影响。DCCT研究中,对1 441位1型糖尿病患者随机分组接受常规或强化血糖控制,时间超过6.5年。并发主要大血管事件的人数在常规组是40人,而在强化治疗组为23人,但这一结果没统计学差异,尽管改善了血脂[3]。
5.2β-受体阻滞剂传统上,不主张用β-受体阻滞剂治疗糖尿病患者,以避免它对胰岛素抵抗的不利作用和发生无意识低血糖。然而随着对心力衰竭和支感神经系统(SNS)在释放血管活性物质方面重要性的进一步认识,它已成为心衰的必要治疗手段。β-受体阻滞剂能够预防甚至逆转心脏重建,改善左心室功能,降低病死率。早期β-受体阻滞剂研究招募的是严重心力衰竭患者,尽管其左心室功能改善,但病死率无降低。CIBIS-Ⅱ(Cardiac Insufficiency Bisoprolol Study Ⅱ)and MERIT-HF (Metoprolol Controlled-release Randomised Intervention Trial in Heart Failure)招募了轻-中度HF患者,病死率分别下降了32%和34%。卡地洛尔,三代β-受体阻滞剂,它能够拮抗α和β受体,已证实能显著地降低发病率和病死率。缺血性心肌患者与无缺血性心肌病患者相比,预后更差;不论是否有糖尿病,均可以从β-受体阻滞剂获益。一项对6个主要β-受体阻滞剂HF试验的荟萃分析显示,糖尿病与病死率升高相关;在试验中接受β-受体阻滞剂的全部患者中,糖尿病患者病死率增加。糖尿病伴有CHF患者用β-受体阻滞剂治疗,其病死率的混合RR与对照组相比是0.84。然而在接受β-受体阻滞剂治疗的非糖尿患者较对照组其相对危险度大为降低[3]。总之β-受体阻滞剂应当在所有的伴有症状HF的糖尿病患者中使用,除非特别情况。它将可以使病死率的RR降低;但其效果没有对非糖尿病患者得到的那么显著; 2组对预后均是明显有益。
5.3ACEI ACEI是心力衰竭治疗的基石。卡托普利多中心研究证实它能够显著改善心衰者的运动能力和症状,但对病死率无改善。CONSENSUS研究小组首先发现依那普利治疗严重心衰患者能显著降低病死率。大量心梗后试验表明,与安慰剂相比,ACEI降低了再发率和病死率。研究发现,雷米普利治疗能够使心血管发病率和死亡率显著获益,而且在糖尿病患者中更为突出[18]。此外,研究还证实了发生新的心衰的危险降低33%,发生2型糖尿病的危险降低44%[18]。
5.4血管紧张素Ⅱ受体拮抗剂血管紧张素Ⅱ是心脏功能障碍的重要参与者。ELITE(evaluation of losartan in the elderly)研究将氯沙坦和卡托普利在老年心衰患者中进行比较,发现氯沙坦和卡托普利安全性相同,尽管其Ⅱ期研究证实氯沙坦的耐受性好于卡托普利,但氯沙坦组的病死率略高[19]。ARBs(angiotensin II type 1 receptor blockers)被证实当它与ACEI联合治疗慢性心衰时,它能产生一个附加效应作用于血液动力学,神经介质活性和左心室重建。Val-HeFT (valsartan heart failure trial)发现,尽管在未用ACEI组可以观察到获益,但在联合应用ACEI和β-受体阻滞剂组却有死亡率升高。
5.5钙通道拮抗剂一项早期的动物研究证明维拉帕米能改善糖尿病性心肌病。然而对维拉帕米,地尔硫卓和硝苯地平的研究指出它们对心衰有不利效应。PRAISE (prospective randomized amlodipine survival evaluation)和Val-HeFTⅢ分别考察了氨氯地平和非洛地平,并未获得超出常规治疗的显著受益,并且非洛地平可使射血分数得到短期改善,但在长期随访时作用未能持续。氨氯地平能够降低致死和非致死事件的发生,减少无局部缺血组死亡危险。PRAISEⅡ正在进行。
5.6他汀类他汀类能够降低血浆胆固醇,减少CHD终点目标已证明了血脂假说的一部分。除对胆固醇代谢的直接作用,他汀类还有很多其他作用,包括修饰GTP结合蛋白如Rho,增加活性斑块下游侧支血流,增强内皮细胞NO合酶的活性,防止AGE诱导的NF-κB-induced protein-1激活,防止VEGF mRNA的增量调节[20]。有研究证实代谢综合征患者发生主要冠脉事件的长期相关危险增加了1.4~1.5倍。而糖耐量下降和糖尿病患者可以获得更佳的效果。HPS (heart protection study)中,5 963位糖尿病患者的血管事件发生降低了22%。而2 912位不伴闭塞性动脉病的糖尿病参与者中事件发生率降低了33%。在2 426位经预处理使LDL胆固醇浓度<3.0 mmol/L的糖尿病患者中,降低了27%。因而不论LDL水平,所有2型糖尿病均可给予他汀类治疗。尽管他汀类在治疗CHD上有显著效果,但他汀类治疗HF这方面却缺少资料。
5.7噻唑烷二酮类(TZDs)TZDs通过结合和激活PPAR-δ(一种能够调控细胞分化的核受体)来增加骨骼肌和脂肪组织的胰岛素敏感性。它们也作用于PPAR-α,增加血浆HDL胆固醇,降低血浆三酰甘油,略微增加LDL胆固醇水平[21]。心肌代谢利用大量的底物生产足够的能量以维持心脏收缩,包括NEFAs,葡萄糖和乳酸。而在2型糖尿病中,由于胰岛素抵抗的结果,葡萄糖未能充分利用,并且NEFA代谢增加了收缩损伤。TZDs除了其增敏作用,它们也增加心脏葡萄糖载体的表达和功能,改善了葡萄糖代谢,并减少了心肌对NEFA的利用。结果是它们保护了缺血所致的心肌损伤,改善了缺血后功能的恢复。
5.8PARP抑制剂PARP-1是PAPP酶家族的一员,当DNA损伤时,PARP可识别结合到DNA断裂处并被激活而参与DNA的修复在内皮细胞,由于高血糖诱使线粒体过氧化物产生过多,使DNA链断裂,激活了PARP,它抑制了GAPDH磷酸甘油醛脱氢酶。导致了葡萄糖蓄积,其他糖酵解中间产物先进入三羧酸循环。激活了高血糖危害的许多主要路径(多元醇途径,糖基化产物形成,PKCβ激活)[22]。PARP也通过调控NF-κB的激活和诱导内皮素-1和受体过度表达来调节心血管炎症和损伤过程。故而,PARP抑制剂阻断PAPP活性,从而阻断了其主要作用路径,减少了对组织的间接损害[23]。
糖尿病性心肌病的发病以能量代谢紊乱为基础,通过多种复杂机制造成微血管的结构和功能改变、心肌细胞的结构和功能改变[24]。进一步深入探讨糖尿病性心肌病的发生机制可以更有效的指导其治疗,而先进诊断技术的发现能早期发现心肌功能改变,早期采取干预措施。
参考文献
1 Debono M,Cachia E.The impact of cardiovascular autonomic neuropathy in diabetes: is it associated with left ventricular dysfunction? Auton Neurosci,2007,132: 1-7.
2 Karamitsos TD,Karvounis HI,Dalamanga EG,et al.Early diastolic impairment of diabetic heart: The significance of right ventricle.Int J Cardiol,2007,114: 218-223.
3 Hayat SA,Patel B,Khattar RS,et al.Diabetic cardiomyopathy: mechanisms,diagnosis and treatment.Clin Sci (Lond),2004,107: 539-557.
4 Palmieri V,Bella JN,Tracy RP,et al.Relationship of leftventricular hypertrophy to inflammation and albuminuria in adults with type 2 diabetes.The Strong Heart Study.Diabetes Care,2003,26: 2764-2769.
5 Farhangkhoee H,Khan ZA,Kaur H,et al.Vascular endothelial dysfunction in diabetic cardiomyopathy: pathogenesis and potential treatment targets.Pharmacol Ther,2006,111: 384-399.
6 Soriano FG,Pacher P,Mabley J,et al.Rapid reversal ofthe diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose)polymerase.Circ Res,2001,89: 684-691.
7 Bidasee KR,Zhang Y,Shao CH,et al.Diabetes increases formation of advanced glycation end products on sarco(endo)plasmic reticulum Ca -ATPase.Diabetes,2004,53: 463-473.
8 Zhou Y,Grayburn P,Karim A,et al.Lipotoxic heart disease in obese rats: implications for human obesity.Proc Natl Acad Sci USA,2000,97: 1794-1789.
9 Way KJ,Katai N,King GL.Proteinkinase C and the development of diabetic vascular complications.Diabet Med,2001,18: 945-959.
10 Fiordaliso F,Li B,Latini R,et al.Myocyte death in streptozotocin-induced diabetes in rats is angiotensin II-dependent.Lab Invest,2000,80: 513-527.
11 Zannad F,Alla F,Dousset B,et al.Limitation of excessive therapy in patients with CHF: insight from the randomized aldactone evaluation study (RALES).Circulation,2000,102: 2700-2706.
12 Richard D,Berra E,Pouyssegur J.Non hypoxic pathways mediate the induction of hypoxia-inducible factor 1α in vascular smooth muscle cells.J Biol Chem,2000,275: 26765-26771.
13 Lee SH,Wolf PL,Escudero R,et al.Early expression of angiogenesis factors in acute myocardial ischemia and infarction.N Engl J Med,2000,342: 626-633.
14 Monkemann H,De Vriese AS,Blom HJ,et al.Early molecular events in the development of the diabetic cardiomyopathy.Amino Acids,2002,23: 331-336.
15 Vinereanu D,Nicolaides E,Boden L,et al.Conduit arterial stiffness is associated with impaired left ventricularsubendocardial function.Heart,2003,89: 449-451.
16 Monteagudo PT,Moises VA,Kohlmann JO,et al.Influence of autonomic neuropathy upon LV dysfunction in insulin-dependent diabetic patients.Clin.Cardiol,2000,23: 371-375.
17 McCormack J,Greenhalgh T.Seeing what you want to see in randomised controlled trials: versions and perversions of UKPDS data.United Kingdom prospective diabetes study.Br Med J,2000,320: 1720-1723.
18 Yusuf S,Gerstein H,Hoogwerf B,et al.HOPE Study investigators Ramipril and the development of diabetes.JAMA,2001,286: 1882-1885.
19 Pitt B,Poole-Wilson PA,Segal R,et al.Effect of Losartan compared with Captopril on mortality in patients with symptomatic heart failure: randomised trial: the Losartan Heart Failure Survival Study ELITE II.Lancet,2000,355: 1582-1587.
20 Callahan AS.Vascular pleiotropy of statins: clinical evidence and biochemical mechanisms.Curr Atheroscler Rep,2003,5: 33-37.
21 Florkowski CM.Management of co-existing diabetes mellitus and dyslipidemia: defining the role of thiazolidinediones.Am J Cardiovasc Drugs,2002,2: 15-21.
22 Kiss L,Szabó C.The pathogenesis of diabetic complications: the role of DNA injury and poly(ADP-ribose)polymerase activation in peroxynitrite-mediated cytotoxicity.Mem Inst Oswaldo Cruz,2005,100: 29-37.
23 Szabó C.Roles of poly(ADP-ribose)polymerase activation in the pathogenesis of diabetes mellitus and its complications.Pharmacol Res,2005,52: 60-71.
24 Marwick TH.Diabetic heart disease.Heart,2006,92: 296-300.
(收稿日期:2014-09-19)
doi:10.3969/j.issn.1002-7386.2015.06.038
【文章编号】1002-7386(2015)06-0903-05
【文献标识码】A
【中图分类号】R 542.2
