在线二维液相色谱-四极杆飞行时间质谱法检测头孢噻吩钠的杂质谱
2015-03-24闻宏亮赵敬丹杨美成
裘 亚, 秦 峰, 闻宏亮, 赵敬丹, 刘 浩, 杨美成
(上海市食品药品检验所, 上海 201203)
技术与应用
在线二维液相色谱-四极杆飞行时间质谱法检测头孢噻吩钠的杂质谱
裘 亚, 秦 峰, 闻宏亮, 赵敬丹, 刘 浩, 杨美成*
(上海市食品药品检验所, 上海 201203)
建立了在线二维液相色谱-四极杆飞行时间质谱检测头孢噻吩钠杂质谱的方法,有效地解决了流动相中含不挥发性磷酸盐的色谱系统不适合用于液相色谱-质谱快速鉴定杂质的难题。一维高效液相色谱(HPLC)以Symmetry C18为色谱柱,以磷酸盐缓冲液(pH 2.5)和乙腈梯度洗脱;二维以ACQUITY UPLC BEH C18为色谱柱,以0.1%(v/v)甲酸水溶液和0.1%(v/v)甲酸乙腈溶液梯度洗脱。以HLB C18为捕集柱,用0.1%(v/v)甲酸水溶液进行捕集和脱盐,采用正离子模式采集数据。对头孢噻吩钠中6个杂质进行了结构鉴定,对其来源进行了分析,并进一步确证了《中国药典》2010年版对头孢噻吩钠杂质A认定有误。采用本方法可以快速、简便、灵敏地对头孢噻吩钠杂质谱进行检测。
二维液相色谱-四极杆飞行时间质谱;杂质谱;头孢噻吩钠
头孢菌素类抗生素是由头孢菌素的母核7-氨基头孢烷酸(7-ACA)接上不同侧链而制成的半合成抗生素,它对细菌的选择作用强,对人几乎没有毒性,具有抗菌谱广、抗菌作用强、耐青霉素酶和过敏反应少等优点,是临床应用广泛的抗生素[1-3]。因其为半合成抗生素,母核由发酵生产,因此杂质较复杂[4]。而杂质是引起过敏、溶血和神经毒性等不良反应的主要因素,故分析和控制其杂质对优化生产过程和提高临床用药安全性至关重要[5,6]。
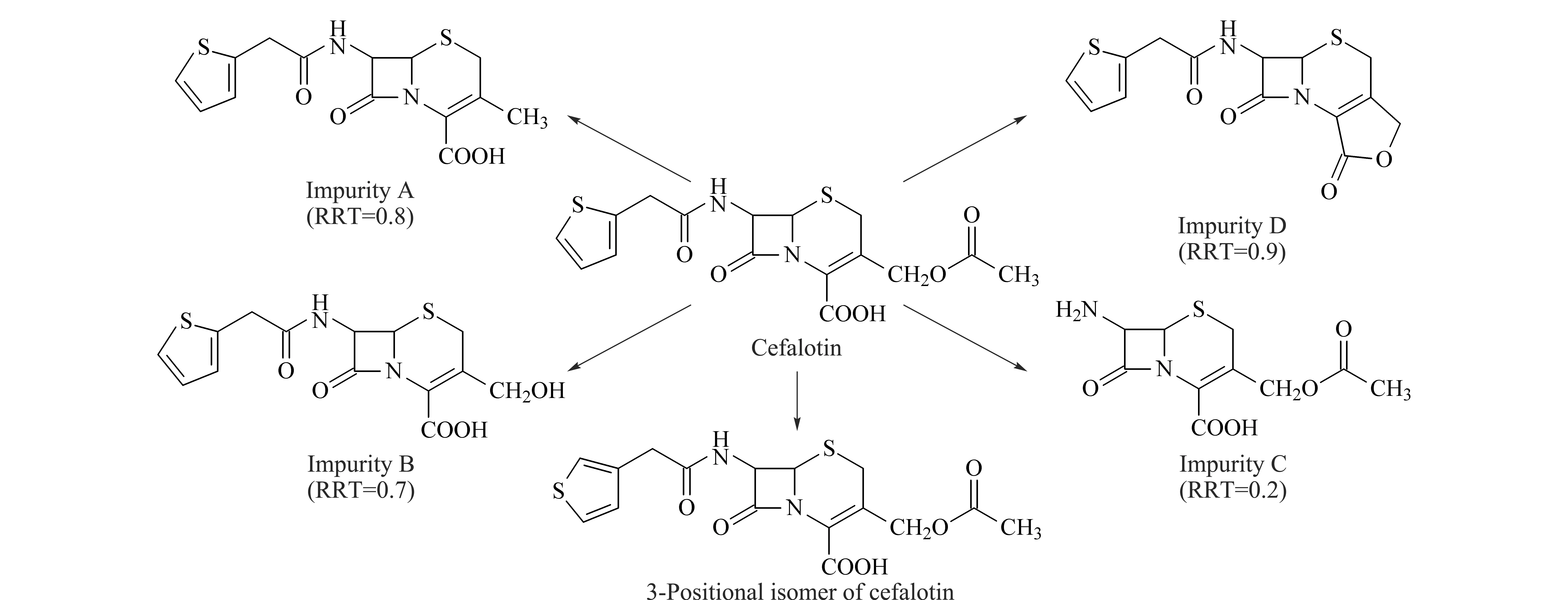
图 1 头孢噻吩钠已知杂质的结构式Fig. 1 Structures of specified impurities of cefalotin sodiumRRT: relative retention time relative to cefalotin sodium.
药品中诸杂质的种类和含量被称为杂质谱,杂质控制已进入“杂质谱控制”阶段[7],对各杂质的定性研究是实现“杂质谱控制”的关键步骤。液相色谱-质谱联用(LC-MS)技术被广泛应用于鉴定药品中的杂质[8-11],但目前头孢菌素类抗生素有关物质分析多采用磷酸盐系统,不适合直接进行LC-MS分析。二维液相色谱-质谱联用技术通过在线联用的自动化操作,可在一维色谱体系中使用质谱不兼容流动相(如磷酸盐系统)对药品进行分析,通过柱切换技术实现对杂质的捕集和脱盐,并转移至二维色谱体系中,使用合适的流动相进行质谱分析[12,13]。
头孢噻吩是第一个也是目前仍广泛应用于临床的头孢菌素类抗生素[14]。临床注射用头孢噻吩钠为头孢噻吩钠的无菌粉末,不含任何辅料。头孢噻吩钠的工业化生产一般由7-ACA与噻吩乙酰氯缩合,再加醋酸钠或2-乙基己酸钠成盐所得[15]。目前对头孢噻吩钠中杂质的检测主要采用HPLC法[15,16]和MEKC (micellar electrokinetic chromatography)法[15,17],有5个杂质已见报道(见图1)。未见使用LC-MS对头孢噻吩钠杂质谱进行检测的报道。《中国药典》2010年版[16]采用磷酸盐流动相进行梯度洗脱的HPLC法检测头孢噻吩钠中的杂质,对其中4个已知杂质(杂质A、B、C和D)采用相对保留时间定位法进行控制,与主成分的相对保留时间如图1所示。本文用在线二维液相色谱-四极杆飞行时间质谱法(2D-LC-QTOF MS)对该磷酸盐系统中头孢噻吩钠的杂质谱进行了分析。对含量(峰面积比)大于0.05%的6个杂质进行了结构鉴定,其中3个为新发现的化合物,分别为头孢噻吩钠工艺副产物和降解产物。进一步确证了《中国药典》2010年版对头孢噻吩钠杂质A认定有误。为头孢噻吩钠的质量控制和工艺改进提供了参考。
1 实验部分
1.1 仪器
ACQUITY UPLC®2D Xevo G2-S QTOF液相色谱-质谱联用仪(美国Waters公司); XS204型电子天平(瑞士梅特勒-托多利公司); Milli-Q超纯水仪(美国Millipore公司)。
1.2 药品与试剂
头孢噻吩钠:生产企业A为苏州中联化学制药有限公司(批号:10110087、10110089、10112026)、生产企业B为上海新亚药业有限公司(批号:100902)、生产企业C为山东罗欣药业股份有限公司(批号:313042102)、生产企业D为齐鲁制药有限公司(批号:2100028AP)、生产企业E为苏州二叶制药有限公司(批号:130304)和Sigma公司(批号:110M0789V);头孢噻吩钠对照品:中国食品药品检定研究院(批号:130407-200707)和欧洲药典委员会(批号:2.0);乙腈和甲酸为色谱级(德国Merck公司);磷酸氢二钾和磷酸为分析级(上海凌峰化学试剂有限公司)。
1.3 溶液的配制
称取头孢噻吩钠75 mg,置于25 mL容量瓶中,加水溶解并稀释至刻度,摇匀。
磷酸盐缓冲液:取磷酸氢二钾1.742 g,加水溶解并稀释至1 000 mL,用磷酸调节pH值至2.5。
1.4 色谱与质谱条件
1.4.1 一维色谱条件
色谱柱:Symmetry C18 (250 mm×4.6 mm, 5 μm);柱温:40 ℃;检测波长:220 nm。流动相A:磷酸盐缓冲液(pH 2.5)-乙腈(97∶3, v/v);流动相B:磷酸盐缓冲液(pH 2.5)-乙腈(3∶2, v/v);进样量:10 μL。梯度洗脱条件:0~30 min,从0%B线性升至100%B,保持到35 min; 35~36 min,从100%B线性降至0%B,并保持到41 min。流速:1.0 mL/min。
1.4.2 二维色谱条件
色谱柱:ACQUITY UPLC BEH C18 (50 mm×2.1 mm, 1.7 μm);柱温:40 ℃。流动相A: 0.1%(v/v)甲酸水溶液;流动相B: 0.1%(v/v)甲酸乙腈溶液。梯度洗脱条件:杂质峰切峰开始后5 min,流动相B由5%升至70%,保持到35 min;35~36 min,从70%B线性降至5%B,并保持到41 min。流速:0.45 mL/min。
1.4.3 捕集条件
溶剂:0.1%(v/v)甲酸水溶液;流速:2 mL/min;捕集柱:HLB C18(30 mm×2.1 mm, 20 μm)。
1.4.4 质谱条件
电离源:ESI+;毛细管电压:1.0 kV;源温度:100 ℃;雾化气温度:500 ℃;雾化气流速:800 L/h;锥孔气流速:50 L/h;质谱软件:MassLynx®4.1。
1.4.5 操作步骤
通过一维紫外检测色谱图确定各杂质的出峰位置,分别对各杂质进行中心切割,通过在液相色谱方法中设置阀切换时间,将杂质捕集至捕集柱上,同时脱盐。再通过阀切换将杂质从捕集柱上反冲至二维色谱柱上分离,最后进入质谱进行分析。四极杆-飞行时间质谱仪采用MSE的采集模式,同时得到一级质谱和二级质谱。使用软件通过精确相对分子质量和同位素分布进行元素组成分析,确定分子式,结合生产工艺和二级质谱碎片推导各杂质可能的结构。
2 结果与讨论
2.1 一维色谱分析
图2为头孢噻吩钠(批号:10110087)样品溶液在一维液相色谱条件下的紫外检测色谱图,其中杂质1~6的含量在0.05%~0.4%之间。本研究分别对这6个杂质进行了结构推导和来源分析。

图 2 头孢噻吩钠样品溶液在一维液相色谱条件下的紫外检测色谱图Fig. 2 First dimensional HPLC-UV chromatogram of cefalotin sodium Peaks 1-6: impurities 1-6.

图 3 杂质4切入二维色谱后的(a)一维紫外检测色谱图和(b)二维总离子流图Fig. 3 (a) First dimensional HPLC-UV chromatogram and (b) the second dimensional total ion current chromatogram after impurity 4 was cut into the second dimensional LCPeaks 1-3, 5, 6: impurities 1-3, 5, 6.
2.2 杂质结构推导
选择含量较低的杂质4进行重点描述。图3为将杂质4(原位于图3a虚线间)从一维液相色谱系统切至二维液相色谱系统后得到的一维紫外检测色谱图和二维总离子流图。二维谱图中保留时间28 min左右的杂质即一维紫外检测色谱图中的杂质4。其一级和二级质谱图如图4所示,根据其精确相对分子质量和同位素分布,推测其分子式可能为C14H13NO6S2。结合其二级质谱碎片及头孢噻吩钠的生产工艺,推测其可能的结构如图5所示。其中m/z232的碎片是侧链在a处发生裂解后所得的母核碎片;m/z186的碎片是同时在a处和b处发生裂解后所得的母核碎片;m/z97的碎片是侧链在c处发生裂解后所得的噻吩亚甲基碎片。

图 4 杂质4的(a)一级和(b)二级质谱图Fig. 4 (a) MS and (b) MS2 spectra of impurity 4表 1 头孢噻吩钠中杂质1~6的MS数据及解析Table 1 MS data and proposed structures of the impurities 1-6 in cefalotin sodium

ImpuritynumberRetentiontime/minRelativeretentiontime*[M+H]+(m/z)KeyfragmentionsofMS2(m/z)MolecularformulaProposedchemicalstructure118.020.70370.9926343,325,297,216,182,156,97C14H11ClN2O4S2218.720.73355.0411337,309,263,226,182,156,97C14H14N2O5S2320.970.82337.0308309,226,182,156,97C14H12N2O4S2421.730.85356.0266338,232,186,158,97C14H13NO6S2524.580.96339.0131309,263,232,182,156,97C14H14N2O4S2631.761.24338.0158279,264,226,199,140,97C14H11NO5S2
* Retention time of the impurity with reference to the retention time of cefalotin.
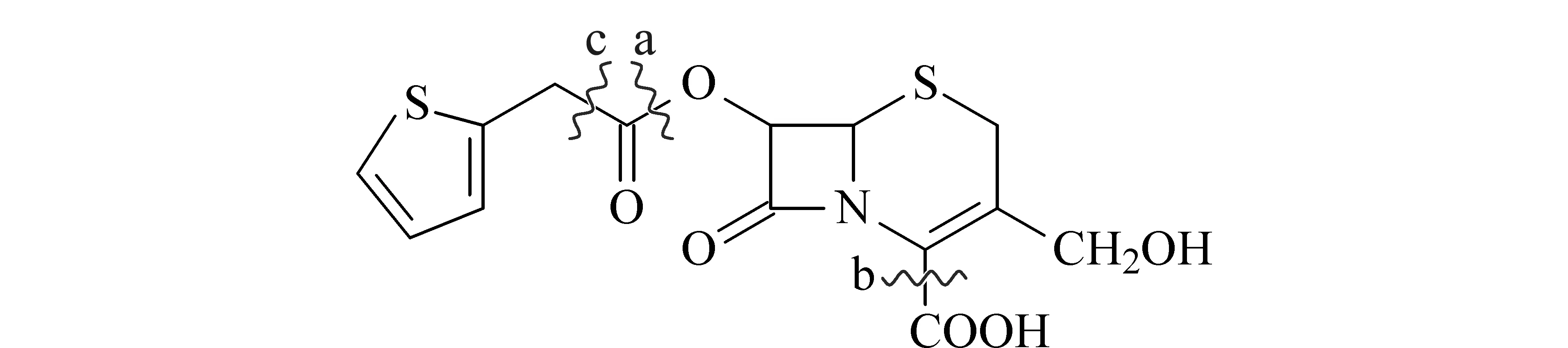
图 5 杂质4可能的结构Fig. 5 Proposed structure of impurity 4
用上述方法分别对其余5个杂质进行分析,其MS数据、分子式和推测所得结构式分别如表1所示。其中,杂质1、杂质4和杂质6为头孢噻吩钠中新发现的杂质;杂质2、杂质3和杂质5分别为头孢噻吩钠已知杂质B、D和A,其相对主峰的相对保留时间分别为0.73、0.82和0.96。其中杂质A的相对保留时间与《中国药典》中规定的相对保留时间(0.8)存在较大差异。该研究结果也与我们之前的研究结果[15]一致,本研究进一步证明《中国药典》2010年版对头孢噻吩钠杂质A认定有误。
2.3 样品测定及杂质来源分析
对不同产地不同批次的头孢噻吩钠样品进行测定,结果见表2(峰面积比即含量小于0.02%的杂质忽略不计)。可见除欧洲药典对照品检出杂质1~5外,其余样品均检出杂质1~6,说明不同来源的头孢噻吩钠的生产工艺类似。
杂质1从来源上分析可能为头孢噻吩钠合成起始原料7-ACA中的杂质去乙酰7-ACA内酯[4,15]与另一起始原料2-噻吩乙酰氯缩合,同时其内酯环上的氢被氯取代后所得的副产物,也可能是降解产生的头孢噻吩钠杂质D内酯环上氢原子被氯取代所得的降解产物。杂质2(即头孢噻吩钠杂质B)可能是7-ACA中的杂质去乙酰7-ACA[4,15]与2-噻吩乙酰氯缩合产生的副产物,也可能是头孢噻吩钠在水溶液中缓慢水解产生的降解产物[15,18]。杂质3(即头孢噻吩钠杂质D)可能是7-ACA中的杂质去乙酰7-ACA内酯[4,15]与2-噻吩乙酰氯缩合产生的副产物,也可能是头孢噻吩钠在弱酸性条件下降解后再环合生成[15,18]。杂质4可能是7-ACA中的氧取代杂质与2-噻吩乙酰氯反应生成的副产物。杂质5(即头孢噻吩钠杂质A)可能是7-ACA中的杂质7-氨基去乙酰氧基头孢烷酸与2-噻吩乙酰氯缩合产生的副产物[15]。杂质6可能是7-ACA中的氧取代杂质与2-噻吩乙酰氯反应生成的副产物。

表 2 不同产地不同批次的头孢噻吩钠样品中杂质的含量Table 2 Contents of the impurities in cefalotin sodium from different manufacturers
3 结论
本研究采用在线二维液相色谱-质谱联用技术对头孢噻吩钠杂质谱进行分析,方法简便、灵敏,有效地解决了流动相中含不挥发性磷酸盐的色谱系统不适合用LC-MS快速鉴定杂质结构的难题。本研究鉴定了头孢噻吩钠中6个杂质(3个已知杂质和3个新发现的杂质)的结构,并结合生产工艺对杂质来源进行了分析,为后续实现对头孢噻吩钠进行“杂质谱控制”奠定了很好的基础,为其他类似的研究提供了较好的研究思路。通过本研究还进一步确证了《中国药典》2010年版对头孢噻吩钠杂质A认定有误。由于杂质结构仅采用LC-QTOF MS手段进行鉴定,信息量仍较少,因此需要进一步对所推断的新杂质进行制备,并采用核磁共振等手段来确证杂质结构。
致谢 感谢Waters公司王志英工程师给予的技术支持。
[1] Salwa R E, Gamal A S, Fardous A M, et al. J Pharm Biomed Anal, 2007, 45: 1
[2] Xue Y, Chen Y Y. Chinese Journal of Antibiotics (薛雨, 陈宇瑛. 中国抗生素杂志), 2011, 36(2): 86
[3] Mo J N, Li W, Wen Y Y, et al. Chinese Pharmaceutical Affairs (莫金娜, 李汶, 温玉莹, 等. 中国药事), 2010, 24(8): 750
[4] Han P, Zhang P P. Drug Standards of China (韩鹏, 张培培. 中国药品标准), 2007, 8(2): 19
[5] Hu M, Hu C Q, Liu W Y. Chinese Journal of Pharmaceutical Analysis (胡敏, 胡昌勤, 刘文英. 药物分析杂志), 2005, 25(3): 369
[6] Hu C Q, Jiang Y, Zhang J P, et al. Chinese Journal of New Drug (胡昌勤, 蒋煜, 张靖溥, 等. 中国新药杂志), 2013, 22(1): 4
[7] Hu C Q. Scientia Sinica Chimica (胡昌勤. 中国科学: 化学), 2010, 40(6): 679
[8] Mallikarjun N, Tarun H, Parul S, et al. J Pharm Biomed Anal, 2014, 87: 191
[9] Ravi N T, Nishit S, Vikas B, et al. J Pharm Anal, 2015, 5(1): 33
[10] Li J, Huang H W, Zhang H, et al. Acta Pharmaceutica Sinica (李婕, 黄海伟, 张红, 等. 药学学报), 2014, 49(5): 672
[11] Song D M, Ren M T, Duan G L. Chinese Journal of Pharmaceutical Analysis (宋东梅, 任美婷, 段更利. 药物分析杂志), 2012, 32(8): 1468
[12] Gao H, Wen X S, Ma X J, et al. Chinese Journal of Pharmaceutical Analysis (高辉, 温学森, 马小军, 等. 药物分析杂志), 2007, 27(4): 616
[13] Wang Z C, Zhang Q H, Zhao Z Y, et al. Chinese Journal of Analytical Chemistry (王智聪, 张庆合, 赵忠一, 等. 分析化学), 2005, 33(5): 722
[14] Yang M C, Qin F, Liu H, et al. Chinese Journal of Pharmaceutical Analysis (杨美成, 秦峰, 刘浩, 等. 药物分析杂志), 2014, 34(8): 1449
[15] Liu H, Jia Z Q. Chinese Journal of Antibiotics (刘浩, 贾志强. 中国抗生素杂志), 2013, 38(12): 921
[16] Pharmacopoeia Commission of People’s Republic of China. Pharmacopoeia of People’s Republic of China, Part 2. Beijing: China Medical Science Press (国家药典委员会. 中华人民共和国药典, 二部. 北京: 中国医药科技出版社), 2010: 215
[17] Yang M C, Liu H, Qiu Y, et al. J Liq Chromatogr Related Technol, 2015, 38(7): 816
[18] Pharmacopoeia Commission of People’s Republic of China. Pharmacopoeia Notes of Pharmacopoeia of People’s Republic of China 1990 Part 2. Beijing: Chemical Industry Press (国家药典委员会. 中华人民共和国药典1990年版二部药典注释. 北京: 化学工业出版社), 1993: 175
Impurity profile study of cefalotin sodium by two-dimensional liquid chromatography-quadrupole time-of-flight mass spectrometry
QIU Ya, QIN Feng, WEN Hongliang, ZHAO Jingdan, LIU Hao, YANG Meicheng*
(ShanghaiInstituteforFoodandDrugControl,Shanghai201203,China)
A two-dimensional liquid chromatography-quadrupole time-of-flight mass spectrometry (2D-LC-QTOF MS) method to profile the impurities of cefalotin sodium was developed. A Symmetry C18 column (250 mm×4.6 mm, 5 μm) was used in the first dimensional chromatography, with gradient elution using pH 2.5 phosphate buffer and acetonitrile as the mobile phases. The column temperature was maintained at 40 ℃ with an ultraviolet detection of 220 nm for analysis. An ACQUITY UPLC BEH C18 column (50 mm×2.1 mm, 1.7 μm) was used in the second dimensional chromatography, with gradient elution using water containing 0.1% (v/v) formic acid and acetonitrile containing 0.1% (v/v) formic acid as the mobile phases. The column temperature was maintained at 40 ℃. An HLB C18 column (30 mm×2.1 mm, 20 μm) was used as the trap column. The data were collected in positive ion mode. The ion source temperature was set at 100 ℃ and the electrospray ionization (ESI) needle voltage was set at 1 000 V. The nebulizer gas temperature was set at 500 ℃. The molecular formulas of the impurities were determined by their exact masses and isotope distributions. And the structures were determined by the protonated molecular ions and the manufacturing process of cefalotin sodium. Six impurities of cefalotin sodium were characterized and the origination of the impurities was deduced. Three of them were unknown impurities to the best of our knowledge. It was confirmed that the Chinese Pharmacopoeia 2010 has mistaken impurity A of cefalotin sodium. The results indicated that the 2D-LC-QTOF MS method could be used to investigate the impurity profile of cefalotin sodium, and it is simple and sensitive.
two-dimensional liquid chromatography-quadrupole time-of-flight mass spectrometry (2D-LC-QTOF MS); impurity profile; cefalotin sodium
cturers A-E; CRS: chemical
ubstance; ChP: Chinese Pharmacopoeia; EP: European Pharmacopoeia.
10.3724/SP.J.1123.2015.07027
中国食品药品检定研究院中青年发展研究基金项目(2013WA4).
2015-07-27
O658
A
1000-8713(2015)12-1314-06
* 通讯联系人.Tel:(021)50798180,E-mail:yangmeicheng@vip.sina.com.
