固相萃取-超快速液相色谱-串联质谱法测定当归中135种农药及其代谢物残留
2015-03-24孟文婷赵云丽于治国
刘 洁, 佟 玲, 孟文婷, 赵云丽, 于治国*
(1. 沈阳药科大学药学院, 辽宁 沈阳 110016; 2. 天士力控股集团研究院药物分析所, 天津 300402)
研究论文
固相萃取-超快速液相色谱-串联质谱法测定当归中135种农药及其代谢物残留
刘 洁1, 佟 玲2, 孟文婷2, 赵云丽1, 于治国1*
(1. 沈阳药科大学药学院, 辽宁 沈阳 110016; 2. 天士力控股集团研究院药物分析所, 天津 300402)
建立了超快速液相色谱-串联质谱(UFLC-MS/MS)同时测定当归药材中135种农药及其代谢物(包括有机磷类、氨基甲酸酯类、拟除虫菊酯类农药等)残留量的分析方法。以回收率为考察指标,评估了不同的提取溶剂、固相萃取小柱、洗脱溶剂及体积对当归中多农残的提取净化效果,最终确定样品经乙腈提取,PSA固相萃取柱净化处理,在电喷雾正离子扫描、依赖保留时间的多反应监测模式(scheduled MRM)下,以基质匹配校准曲线内标法定量。结果表明,135种农药及其代谢物在各自的浓度范围内线性关系良好(r>0.99); 3个添加水平(10、50、100 μg/kg)下,除了烯草酮回收率偏低(62.0%~68.2%)外,其余农药的回收率为71.3%~119.7%,相对标准偏差(RSD,n=6)不大于19.9%, 135种农药及其代谢物的定量限为1.0~10.0 μg/kg。该方法样品前处理简单、快速、灵敏,可用于当归药材中多类别农药残留量的定性、定量。
高效液相色谱-串联质谱;固相萃取;多类别农药及其代谢物;当归
当归(Angelicasinensis(Oliv.)Diels)为伞形科(Umbelliferae)当归属多年生草本药用植物。现代药理和临床研究表明,当归对多种疾病特别是心血管系统疾病有很好的疗效,同时具有抗肿瘤、改善肺功能、提高免疫力等药理活性。近年来由于当归种植面积扩大,病害逐年加重[1]。药农为保证产量在种植过程中使用大量农药以防病虫害,如使用呋喃丹、吡虫啉、毒死蜱、多菌灵等农药防治麻口病等病害[2,3],利用除草剂扑草净解决除草难问题等[4]。当归病虫害防治中允许使用的有机合成农药包括杀虫剂、杀螨剂、杀菌剂、除草剂,而此类农药只允许在中药材生产上限量使用。因此过量使用农药易引发残留超标问题。农药残留量的超标不仅危害人类的健康、损害消费者的利益,也是制约中药走向国际市场的瓶颈。欧盟、美国、日本及国际食品法典委员会(CAC)等国家和组织[5-7]对部分中草药中的有机磷农药、氨基甲酸酯类、拟除虫菊酯类等农药均制定了最高残留限量(MRL)。因此,建立当归药材中多类别农药残留的检测方法,对保证用药安全及推进中药现代化、国际化具有重要的意义。
农药多残留分析需解决的主要问题之一是去除共提取基质干扰物对定量准确度、灵敏度及仪器污染的影响。因此,样品前处理中对基质净化方法的选择及优化是多残留检测的关键。固相萃取技术具有吸附容量大、净化效果好、易与检测仪器联用等优点,已广泛应用于中药材农药多残留分析[8-10],是当前国内实验室普遍采用的前处理方法。
传统的仪器检测方法,如气相色谱法(GC)、液相色谱法(HPLC),难以消除复杂基质的干扰。气相色谱-质谱联用法(GC-MS)虽在中药材农药残留检测方面做出很大的贡献,如程志等[11]采用GC-MS快速筛查了11味中药材中的144种农药。但GC-MS常常会受到化合物极性及处理过程相对复杂、耗时等限制[10],且其定量限一般较高,难以满足国际社会对农药残留限量日益严格的检测要求[12]。液相色谱-质谱联用技术(LC-MSn)凭借快速、抗干扰能力强、应用范围广等优势,已成为痕量定量分析的首选方法[13]。
本文对固相萃取前处理方法中提取和净化步骤进行了优化和评估,结合灵敏度高、抗干扰能力强的LC-MS/MS技术,以基质匹配校准曲线内标法一次进样即可完成当归药材中多类别农药残留的定性、定量分析,并应用于实际样品的分析检测。该方法具有操作简单、灵敏度高、应用广的特点,对当归药材的安全监测有重要的意义。
1 实验部分
1.1 仪器、试剂与材料
QTRAP 5500质谱仪,配有电喷雾离子源、三重四极杆-线性离子阱质量分析器及Analyst 1.6.2工作站(美国AB SCIEX公司)、MultiQuant 3.0.2数据处理软件;LC-30AD UFLC液相色谱系统(日本岛津公司)。
乙腈为色谱纯(德国Merck公司);无水硫酸镁为优级纯(Sigma-Aldrich);醋酸钠为分析纯(天津市化学试剂一厂); PSA固相萃取柱(500 mg/6 mL)、CARB固相萃取柱(500 mg/6 mL)、CARB/NH2固相萃取柱(250 mg/250 mg/6 mL)、C18固相萃取柱(500 mg/6 mL)、Florisil固相萃取柱(500 mg/6 mL)和NH2固相萃取柱(500 mg/6 mL)均为DIKMA科技公司生产。实验用水为Milli-Q纯水系统所制超纯水。
135种农药及其代谢物、内标d5-莠去津标准品(纯度≥95%),购自德国Dr. Ehrenstorfer公司与中国农业部环境保护科研监测所。
中药材样品(10批当归),产地为甘肃,由天津天士力中药资源有限公司提供。
1.2 标准溶液的制备
单标准储备液:取各农药标准品10 mg(精确至0.001 mg),分置于10 mL量瓶中,用乙腈溶解并稀释至刻度,制成质量浓度为1.0 g/L的单标准储备液,储存于4 ℃冰箱中,备用。
混合储备液:分别准确吸取适量单标准储备液,用0.05%(v/v)乙酸乙腈溶液定量稀释,制成各农药组分质量浓度均为1.0 mg/L的混合储备液,于-20 ℃冰箱中保存。
内标溶液:取d5-莠去津标准品10 mg(精确至0.001 mg),置于10 mL量瓶中,用乙腈溶解并稀释至刻度,制成质量浓度为1.0 g/L的内标储备液;准确吸取内标储备液适量,用乙腈定量稀释,制成质量浓度为5 mg/L的内标溶液,储存于4 ℃冰箱中,备用。
标准工作溶液:准确吸取系列体积的混合储备液,分置于5 mL量瓶中,各加入内标溶液50 μL,用乙腈稀释至刻度,制成质量浓度分别为1、2、5、10、20、50、80 μg/L的标准工作溶液。
基质匹配标准溶液:按照1.3节步骤提取不含目标化合物的“空白”样品,将获得的提取液作为稀释溶剂,按照上述“标准工作溶液”的配制,制成相同浓度梯度的基质匹配标准溶液。
1.3 样品前处理
称取当归药材粉末2 g(精确至0.01 g),置于50 mL离心管中,准确加入内标溶液100 μL、水10 mL,浸润30 min后,准确加入乙腈10 mL,于涡旋混合振荡仪振荡(5 000 r/min)2 min,向离心管中加入无水硫酸镁4 g和醋酸钠1 g,手动振摇5 min,离心(5 000 r/min)5 min。
取上清液1 mL,加至预先用乙腈-甲苯(20∶1, v/v)5 mL活化的固相萃取小柱中,用乙腈-甲苯(20∶1, v/v)20 mL分4次淋洗小柱,合并洗脱液,在38 ℃水浴中旋转蒸发至近干,用乙腈1 mL溶解残渣,经0. 22 μm滤膜过滤,滤液作为供试溶液。
1.4 UFLC-MS/MS条件
1.4.1 UFLC条件
色谱柱:Acquity HSS T3柱(100 mm×2.1 mm, 1.8 μm,Waters公司);流动相:0.1%(v/v)甲酸水溶液(A)和乙腈(B);流速:0.3 mL/min;梯度洗脱程序:0~1.0 min, 95%A; 1.0~4.0 min, 95%A~40%A; 4.0~14.0 min, 40%A~0%A; 14.0~15.0 min, 0%A; 15.0~18.0 min, 95%A。
1.4.2 MS/MS条件
离子源:电喷雾离子(ESI)源;扫描方式:正离子扫描;检测方式:依赖保留时间的多反应监测(scheduled MRM)模式;离子化电压(IS): 5 500 V;雾化温度(TEM): 550 ℃;气帘气压力(CUR): 241.3 kPa (35 psi);喷雾气压力(Gas 1): 379.3 kPa (55 psi);辅助加热气压力(Gas 2): 379.3 kPa (55 psi);碰撞气压力(CAD):中等(medium)。
2 结果与讨论
2.1 农药种类的选择
本研究根据农药的理化性质,依据GB/T 20769-2008,并参考美国、欧盟等国家和组织[14,15]对植物药中有关农药残留检测项目,筛选了中药材生产中常用的135种不适合直接采用气相色谱和气相色谱-质谱联用法测定的农药及其代谢物。
2.2 UFLC-MS/MS条件的优化
采用MS-only模式,直接进样方式,对单一化合物逐个进行优化。首先进行Q1全扫描,识别目标物的母离子,参考欧盟指令(2002/657/EC)[16],对该母离子进行Q2扫描。每个化合物选择2对响应值高的特征离子对作为定量与定性离子对。各农药的质谱参数如表1所示。应用scheduled MRM扫描方式,可确保每个色谱峰有足够的采集数据点(每个色谱峰点数在12~20之间)。
为了保证各农药化合物具有良好峰形以及足够的灵敏度,本文考察了乙腈-水、乙腈-0.1%(v/v)甲酸水溶液、乙腈-0.1%(v/v)甲酸水溶液(含5 mmol/L甲酸铵)以及乙腈-0.1%(v/v)甲酸水溶液(含10 mmol/L甲酸铵)4种流动相体系。甲酸的加入,可促进化合物的离子化[12]。因此,当流动相中加入0.1%(v/v)甲酸时,各化合物的响应明显增强。盐的加入,不仅可影响化合物的保留能力、峰形,也可增强或抑制化合物的离子化作用。分别尝试加入5 mmol/L和10 mmol/L甲酸铵,发现随着盐浓度的增加,化合物的响应逐渐减弱,说明盐对化合物的电离产生抑制作用。依据电喷雾电离的机理,选择有利于化合物去质子化的乙腈为有机相[10]。因此本文以乙腈-0.1%(v/v)甲酸水溶液为流动相。

表 1 135种农药及其代谢物的保留时间及质谱分析参数Table 1 Retention times and mass spectrometric parameters for the 135 pesticides and their metabolites

表 1 (续)Table 1 (Continued)

表 1 (续)Table 1 (Continued)

表 1 (续)Table 1 (Continued)
* for quantitative ion; ** qualitative ion response/quantitative ion response×100%; *** internal standard.

图 1 使用不同提取溶剂时当归提取液的总离子流色谱图Fig. 1 Total ion chromatograms of Angelica sinensis extract with different extraction solvents
2.3 样品前处理方法的优化
2.3.1 水用量的确定
由于中药材含水量较少,直接使用有机溶剂难以有效提取待分析物。因此需加入一定量的水,以确保样品中的待分析物较容易地被提取出来。称取药材2 g,分别加入水5、10和15 mL,静置30 min。结果显示,5 mL水不能使药材完全浸润,而15 mL水量过多,影响液-液分配效率,降低回收率。因此,最终确定中药材取样量为2 g,添加水量为10 mL。
2.3.2 提取溶剂的选择
农药残留检测前处理中常用的提取剂有乙腈、丙酮、乙酸乙酯等,本文分别从净化效果、提取回收率两方面考察了这3种溶剂对135种农药及其代谢物的提取情况。从净化效果(见图1)看,乙酸乙酯由于对水的溶解度较低,极性干扰物不易被同时提取;丙酮极性较大,且与水互溶,易将水溶性的极性干扰物(如色素)一并提取出来,使后续的净化步骤难度加大,导致其重复性较差;3者的净化效果依次为乙酸乙酯>乙腈>丙酮。对135种农药及其代谢物在0.1 mg/kg浓度水平下进行了添加回收试验,图2显示,乙酸乙酯无法从含水基质中完全萃取出极性较大的农药;与丙酮和乙酸乙酯比较,乙腈适合的农药极性范围相对广泛,是一种较为适宜的提取溶剂;3者的提取效率依次为乙腈>丙酮>乙酸乙酯。综合考虑,本实验选择乙腈为提取溶剂。

图 2 不同溶剂提取当归中135种农药及其代谢物的回收率范围(n=3)Fig. 2 Recovery ranges of the 135 pesticides and their metabolites in the Angelica sinensis with different extraction solvents (n=3)表 2 采用不同盐析剂时当归中135种农药及其 代谢物的回收率范围Table 2 Recovery ranges of the 135 pesticides and their metabolites in the Angelica sinensis with different salting-out agents
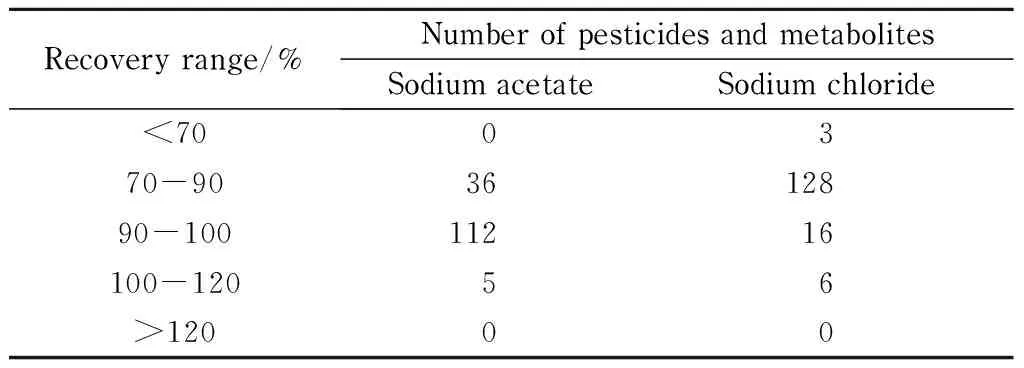
Recoveryrange/%NumberofpesticidesandmetabolitesSodiumacetateSodiumchloride<700370-903612890-10011216100-12056>12000
2.3.3 脱水剂与盐析剂的考察
在提取液中加入硫酸镁作为脱水剂能有效减小水相的体积,从而促进分配于水相的极性分析物再分配进入有机相,以获得高回收率。但是同时考虑到提取的选择性,在采用硫酸镁分离的同时,通过改变添加到样品中的盐,可以控制该方法的极性范围[17],而且可以降低提取溶剂的水溶性,进而减小水溶性基质成分的共提取、降低基质效应。常用的盐析剂有氯化钠、乙酸钠等。本文比较了乙酸钠与氯化钠的提取效率。结果显示,农药在乙酸钠体系中的回收率普遍高于氯化钠体系(见表2)。经测定,乙酸钠体系的pH为5~6,而氯化钠体系的pH为4~5,可见乙酸钠体系抗基质自身酸碱度干扰的能力强,有利于农药的稳定,尤其是pH敏感型农药,如甲苯氟磺胺、灭螨蜢、咪酰胺等。因此本文采用无水硫酸镁、乙酸钠分别作为脱水剂和盐析剂。
2.3.4 SPE柱的选择
固相萃取方法中常用的SPE小柱有PSA、NH2、CARB、C18固相萃取柱等。本文考察了不同填料的固相萃取柱对135种农药及其代谢物的提取回收率影响。如图3所示,使用PSA固相萃取柱时,135种农药及其代谢物的回收率在70%~120%范围内的数目最多(143个),占总数的93%。通过净化后收集液的颜色评价不同小柱的净化效果,试验中发现,净化效果依次为Florisil>CARB>CARB/NH2>PSA>NH2>C18。Florisil为应用最早的填料,由于这种填料会吸附极性强的有机磷农药,因此不适用于本文的多残留分析。CARB虽然可有效除去基质中大量的色素成分,但不能消除基质中的色谱共流出物干扰,不利于减少基质噪声信号[18]。PSA由硅胶键合N-丙基-乙二胺得到,有两个氨基,与NH2柱相似,但比NH2柱具有更强的离子交换能力。因此PSA柱通过极性吸附或弱阴离子交换作用,实现了对中药基质中的色素、有机酸、脂肪酸和强阴离子等多种组分的净化;同时还可以保证农药具有较高的回收率。本文最终采用PSA固相萃取柱净化。
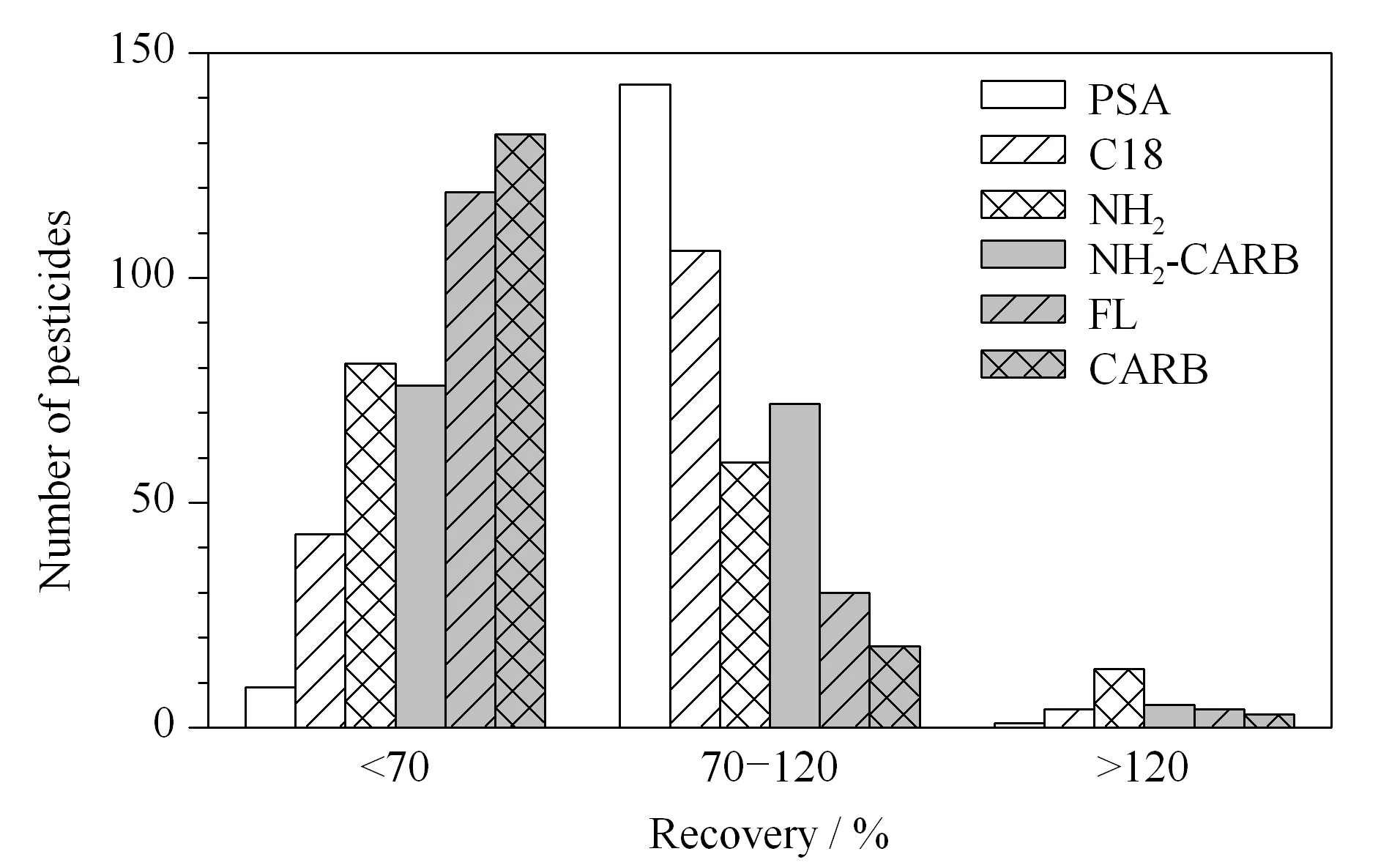
图 3 不同固相萃取柱对当归药材中135种农药及其代谢物回收率的影响(n=3)Fig. 3 Effect of the different SPE cartridges on recoveries of the 135 pesticides and their metabolites in the Angelica sinensis (n=3)
2.3.5 洗脱溶剂及洗脱体积的选择
[19],考察了以乙腈、不同比例的乙腈-甲苯(20∶1、10∶1、8∶1、5∶1、3∶1, v/v)及不同比例的乙腈-乙酸乙酯(3∶1、5∶1, v/v)作为洗脱溶剂对农药回收率的影响。图4表明:乙腈-甲苯比例为20∶1时,回收率在70%~120%范围内的农药及代谢物比例最高,占总数的89%;乙腈为洗脱溶剂时,由于极性较大,共流出干扰物的增多导致农药回收率降低;随着甲苯比例的增加,洗脱溶剂的极性减小,洗脱能力减弱,使得极性农药(甲胺磷、乙酰甲胺磷、氧乐果等)不易被完全洗脱;乙酸乙酯作为极性调节剂的效果没有甲苯好。此外,甲苯毒性较大,应尽量减少甲苯的添加量。本文同时对洗脱体积(10、15、20、25 mL)进行了考察,随着洗脱体积的增加,135种农药及其代谢物的平均回收率升高,当洗脱体积为20 mL时,平均回收率最高,继续增加洗脱量,平均回收率没有显著变化,而共洗脱出的杂质相应增多。因此,最终确定采用20 mL乙腈-甲苯(20∶1, v/v)洗脱PSA固相萃取柱。

图 4 不同洗脱溶剂对当归药材中135种农药及其代谢物回收率的影响(n=3)Fig. 4 Effect of the different elution solvents on recoveries of the 135 pesticides and their metabolites in the Angelica sinensis (n=3) A∶T: acetonitrile∶toluene; A∶E: acetonitrile∶ethyl acetate.
2.3.6 浓缩温度的考察
实验中发现,洗脱液的浓缩温度是影响热不稳定农药回收率的主要因素,如敌百虫在一定温度下会降解生成三氯乙醛、磷酸二甲酯和敌敌畏[20];烯草酮为环己烯酮类除草剂,受热易被氧化。考察了在35、38、40 ℃下旋转蒸发浓缩对个别农药回收率的影响,结果表明,40 ℃时,旋蒸时间缩短,但热不稳定农药烯草酮回收率仅44.3%,氨基甲酸酯类农药呋线威的回收率仅51.9%,有机磷农药磷胺、甲胺磷、敌百虫回收率分别为65%、63%和69%。在35 ℃和38 ℃下,除烯草酮(回收率66%、63%)外,其余农药回收率均在70%以上。为保证较短的前处理时间,最终采用在38 ℃下旋蒸浓缩。
2.4 方法学验证
2.4.1 线性范围与定量限
将1.2节配制的基质匹配标准溶液进样,以各组分定量离子的峰面积与内标峰面积的比值(y)对各组分质量浓度(x, μg/L)绘制标准曲线,采用最小二乘法得回归方程y=a+bx。135种农药及其代谢物在当归药材基质中的响应值与其浓度均呈良好的线性关系,在相应的浓度范围之内,135种农药及其代谢物的线性相关系数(r)均大于0.99。
定量限(LOQ)是指在可接受的准确度和精密度下,能够定量测定的样品中待测成分的最低量或浓度[21]。本实验通过向当归空白基质中添加多个较低水平的农药混合标准溶液分别进行基质加标试验,每个添加水平各平行测定6次,计算平均回收率和相对标准偏差(RSD),回收率介于70%~120%及RSD小于20%,同时满足信噪比(S/N)≥10的最低添加水平即该物质的定量限。135种农药及其代谢物定量限均在1~10 μg/kg之间,低于各国及组织的法规对农药残留限量的要求。最低添加水平下定量稀释,以信噪比(S/N)≥3时的浓度为检出限(LOD),为0.3~3 μg/kg。结果见表3。
2.4.2 基质效应
基质效应(matrix effect, ME)是指样品中除分析物以外的组分对分析物的离子化产生增强或抑制作用,ME影响分析结果的准确性。本文采用以下公式评价ME:


图 5 135种农药及其代谢物在当归中的基质效应分布Fig. 5 Distribution of matrix effects of the 135 pesticidesand their metabolites in Angelica Sinensis
试验发现,不同农药在当归基质中的ME不同。如图5所示,50多个农药在当归药材中产生明显的基质抑制效应(ME<70%)。用以补偿ME的方法有:基质净化法、同位素内标法、空白基质配制标准溶液和加入分析保护剂等。内标法不仅可以对ME进行校正、保证回收率,而且可对样品处理过程中的变化进行校正,有利于方法的稳定性。但对多种类农药同时检测时,由于存在极性差异,即使是同类物质的同位素内标也很难抵消ME,造成定量结果偏差。而基质匹配标准溶液法是按照样品前处理步骤提取不含目标化合物的“空白”样品,将提取液作为溶剂,配制成标准工作溶液。因此,在LC-MS/MS多残留分析中采用基质匹配标准曲线-内标法进行校准定量能较好地补偿ME。

表 3 135种农药及其代谢物的定量限、检出限、相关系数、线性范围、加标回收率及精密度Table 3 LOQs, LODs, correlation coefficients (r), linear ranges, spiked recoveries and relative standard deviations of the 135 pesticides and their metabolites

表 3 (续)Table 3 (Continued)
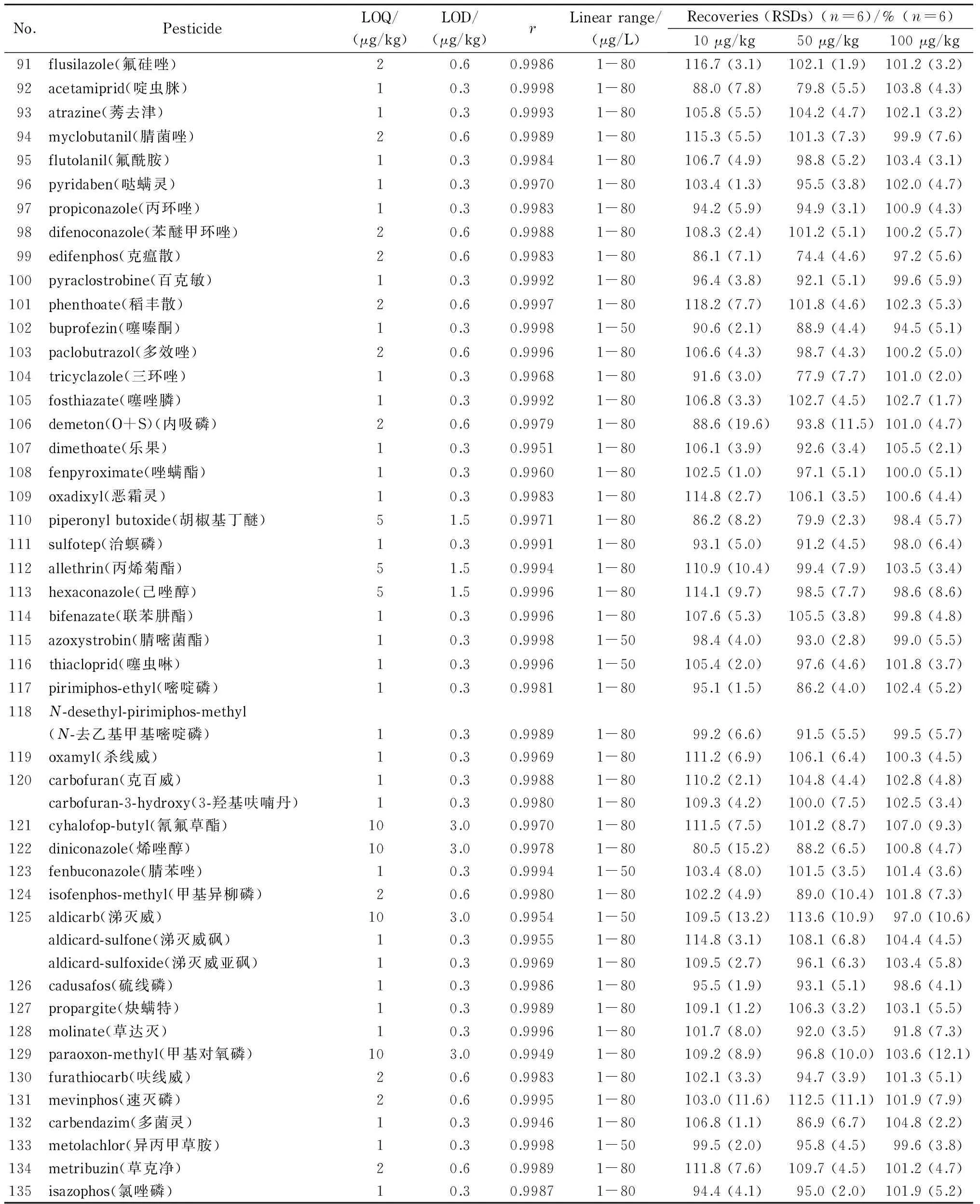
表 3 (续)Table 3 (Continued)
2.4.3 准确度与精密度
本文采用基质匹配标准曲线-内标法定量,在无农药残留的当归空白基质中添加135种农药及其代谢物进行回收率试验。准确称取药材粉末2 g,置于50 mL离心管中,添加水平分别为10、50和100 μg/kg,按照1.3节所述的步骤进行样品前处理,每个添加水平重复6次。3个添加水平下,除了烯草酮回收率偏低(62.0%~68.2%),其余农药的平均回收率为71.3%~119.7%, RSD为1.0%~19.9%,数据表明该方法的精密度和回收率都比较好,符合残留检测有关标准和法规的要求。
2.5 定性依据
参考GB/T 20769-2008,阳性结果的确证依据为:样品中待测物的保留时间与相应标准品的保留时间偏差在±0.2 min以内;同时样品中各农药定性离子的相对丰度与相应标准品定性离子的相对丰度必须符合下述标准:相对丰度大于50%,偏差在±10%;相对丰度为20%~50%,偏差在±20%;相对丰度小于10%,偏差在±50%。
2.6 实际样品的测定
应用所建立的分析方法对10批当归样品进行农药的快速筛查。根据2.5节所述定性依据,以保留时间、离子对比率进行定性分析,以定量离子对进行定量分析。在6批药材中检出农药残留,检出率高达60%。检出的农药有多菌灵、甲拌磷、莠去津、烯唑醇,含量依次为0.010~0.020 mg/kg、0.018~0.051 mg/kg、0.012~0.018 mg/kg、0.015~0.049 mg/kg,其中甲拌磷和烯唑醇超出欧盟限量要求(MRL为0.01 mg/kg),但低于CAC的规定(MRL为0.1 mg/kg)。分析结果发现不同种类农药在当归药材中有不同程度的残留,这可能由于药材在种植、生产、储存过程中接触不同种类农药有关。
3 结论
本文建立了当归药材中135种农药及其代谢物残留快速检测的SPE-UFLC-MS/MS方法。该方法操作简单、净化效果好、灵敏度高,为当归药材中多类别农药残留的例行检测、风险评估等研究提供了一种高效、可靠的分析手段。
参考文献:
[1] Bian J, Chen T X, Chen X R, et al. Acta Prataculturae Sinica (卞静, 陈泰祥, 陈秀蓉, 等. 草业学报), 2014, 23(6): 266
[2] Kong J, Li J G, Wang J, et al. Journal of Northwest Normal University (孔杰, 李建刚, 王剑, 等. 西北师范学院学报), 1988(3): 50
[3] Cui J J, Song X B, Ma X J, et al. Modern Chinese Medicine (崔建军, 宋学斌, 马晓嘉, 等. 中国现代中药), 2012, 14(6): 28
[4] Wang L. [MS Dissertation]. Yanji: Yanbian University (王亮. [硕士学位论文]. 延吉: 延边大学), 2007
[5] Zhuang W J. The Global Regulations on Maximum Residue Limits (MRLs) for Pesticides in Foodstuffs and Feedstuffs: Volumes Ⅰ-Ⅳ. Beijing: Chemical Industry Press (庄无忌. 国际食品饲料中农药残留限量法规: Ⅰ~Ⅳ卷. 北京: 化学工业出版社), 2010
[6] GB 2763-2014
[7] Lin W X. The Compilation of Residue Limit Standards for Pesticides and Veterinary Drugs in Foodstuffs in the World. Dalian: Dalian Maritime University Press (林维宣. 各国食品中农药兽药残留限量规定. 大连: 大连海事大学出版社), 2002
[8] Zhang Y, Zhang Q, Wang X Q. Chinese Journal of Pharmaceutical Analysis (张园, 张琦, 王贤亲. 药物分析杂志), 2012, 32(1): 95
[9] Ding M, Zhong D L, Tang F B, et al. Chinese Journal of Chromatography (丁明, 钟冬莲, 汤富彬, 等. 色谱), 2013, 31(2): 117
[10] Li N, Shao H, Liu L, et al. Chinese Journal of Pharmaceutical Analysis (李娜, 邵辉, 刘磊, 等. 药物分析杂志), 2012, 32(5): 852
[11] Cheng Z, Zhang R, Liu W H, et al. Chinese Journal of Chromatography (程志, 张蓉, 刘韦华, 等. 色谱), 2014, 32(1): 57
[12] Wang F, Li T, Ma C. Chinese Journal of Chromatography (王菲, 李彤, 马辰. 色谱), 2013, 31(3): 191
[13] Wang L Z, Zhou Y, Huang X Y, et al. Chinese Journal of Chromatography (王连珠, 周昱, 黄小燕, 等. 色谱), 2013, 31(12): 1167
[14] European Pharmacopeial Convention. European Pharmacopoeia 7.0. (2012-06-01). http://db.yaozh.com/foreign/EP7.0/20813E.PDF
[15] The United Stated Pharmacopeial Convention. United States Pharmacopeia 34. (2011-04-24). http://ishare.iask.sina.com.cn/f/14853792.html
[16] European Commission Decision 2002/657/EC
[17] Mei T, Wu R S, Wei B. China Fruit Vegetable (梅婷, 吴荣顺, 魏波. 中国果菜), 2014, 34(2): 62
[18] Wang Y J, Huang H L, Zhuo H H, et al. Journal of Inspection and Quarantine (王玉健, 黄惠玲, 卓海华, 等. 检验检疫学刊), 2009, 19(2): 74
[19] Jin C L. [MS Dissertation]. Baoding: Hebei University (金春丽. [硕士学位论文]. 保定: 河北大学), 2012
[20] Yu H J, Cai Y Q, Li Q, et al. Chinese Journal of Chromatography (于惠娟, 蔡友琼, 李庆, 等. 色谱), 2006, 24(1): 23
[21] Wang J H, Xu W Y, Sun L, et al. Chinese Journal of Pharmaceutical Analysis (王京辉, 许玮仪, 孙磊, 等. 药物分析杂志), 2012, 32(9): 1704
Determination of 135 pesticides and their metabolites inAngelicasinensisby ultra-fast liquid chromatography-tandem mass spectrometry coupled with solid-phase extraction
LIU Jie1, TONG Ling2, MENG Wenting2, ZHAO Yunli1, YU Zhiguo1*
(1.SchoolofPharmacy,ShenyangPharmaceuticalUniversity,Shenyang110016,China;2.PharmaceuticalAnalysisInstitute,TaslyHoldingGroupAcademy,Tianjin300402,China)
A method using solid-phase extraction (SPE) followed by ultra-fast liquid chromatography-tandem mass spectrometry (UFLC-MS/MS) has been established for simultaneously quantitative determination of 135 pesticides and their metabolites (organophosphorus pesticides, pyrethroid pesticides and carbamate pesticides, etc.) inAngelicasinensis. The pesticide residues were extracted from the samples by acetonitrile, cleaned-up with a primary secondary amine (PSA) column and then analyzed using UFLC-MS/MS in multiple reaction monitoring (MRM) mode with positive electrospray ionization. The pesticide residues were quantified by matrix matched standard solution-internal standard method. All of the pesticides had good linear responses withr>0.99. The average recoveries of the pesticides at the spiked levels of 10, 50, 100 μg/kg ranged from 71.3%-119.7% with the RSDs of 1.0%-19.9%, except for clethodim (62.0%-68.2%). The limits of quantification of the 135 pesticides and their metabolites were 1.0-10.0 μg/kg. The results demonstrated that the method is simple, fast, sensitive and can be used for the analysis of the multiclass of pesticide residues inAngelicasinensis.
ultra-fast liquid chromatography-tandem mass spectrometry (UFLC-MS/MS); solid-phase extraction (SPE); multiclass of pesticide residues and metabolites;Angelicasinensis
10.3724/SP.J.1123.2015.09014
国家“重大新药创制”科技重大专项(2013ZX09402202).
2015-09-14
O658
A
1000-8713(2015)12-1257-12
* 通讯联系人.Tel:(024)23986295,E-mail:zhiguo-yu@163.com.
