海洋放线菌代谢产物、非核糖体多肽、腺苷化结构域研究进展
2015-03-23陆胜利
陆胜利, 祁 超
(1.安庆医药高等专科学校 药学系, 安徽 安庆 246052; 2.华中师范大学 生命科学学院, 武汉 430079)
海洋放线菌代谢产物、非核糖体多肽、腺苷化结构域研究进展
陆胜利1, 祁 超2*
(1.安庆医药高等专科学校 药学系, 安徽 安庆 246052; 2.华中师范大学 生命科学学院, 武汉 430079)
概述了海洋放线菌代谢产物、非核糖体多肽、腺苷化结构域的研究进展.
海洋放线菌; 代谢产物; 腺苷化结构域
1 放线菌概述
放线菌是一类主要呈菌丝状生长和以孢子繁殖的高(G+C)%革兰阳性细菌,属厚壁菌门、放线细菌纲.土壤中的放线菌在死去动植物降解和腐殖质形成中发挥关键作用,它们能利用诸如角质素、木质纤维素和几丁质这类不易降解的多聚物作为营养物来源,使自然界生物物质的再循环得以实现.
放线菌具有丰富的生理学和形态学多样性,在自然界的分布十分广泛,主要以孢子或菌丝状态存在于土壤、空气和水中,尤其是在含水量低、有机物丰富、呈中性或微碱性的土壤中数量最多,可达105~106个/克,土壤特有的泥腥味也是因放线菌产生的土腥味素所致.放线菌目可分为10个亚类,其代表菌属主要有链霉菌属(Streptomyces)、诺卡氏菌属(Nocardia)、放线菌属(Actinomyces)、小单孢菌属(Micromonospora)、孢囊链菌属(Streptosporangium)和游动放线菌属(Actinoplanes)等(见图1).
放线菌与人类的生产、生活有着极为密切的关系,其天然代谢产物一直是现代医药业、农业和畜牧业中药物先导化合物的一个主要来源.现已发现的大约23 000种微生物活性次级代谢产物中,45%以上(超过10 000种)都来自放线菌家族,这其中包括了约70%的已知抗生素[1].药物研发中,既直接提取放线菌的天然活性产物作药物成分,也以其作为母体或模板进行化学修饰合成或全合成.
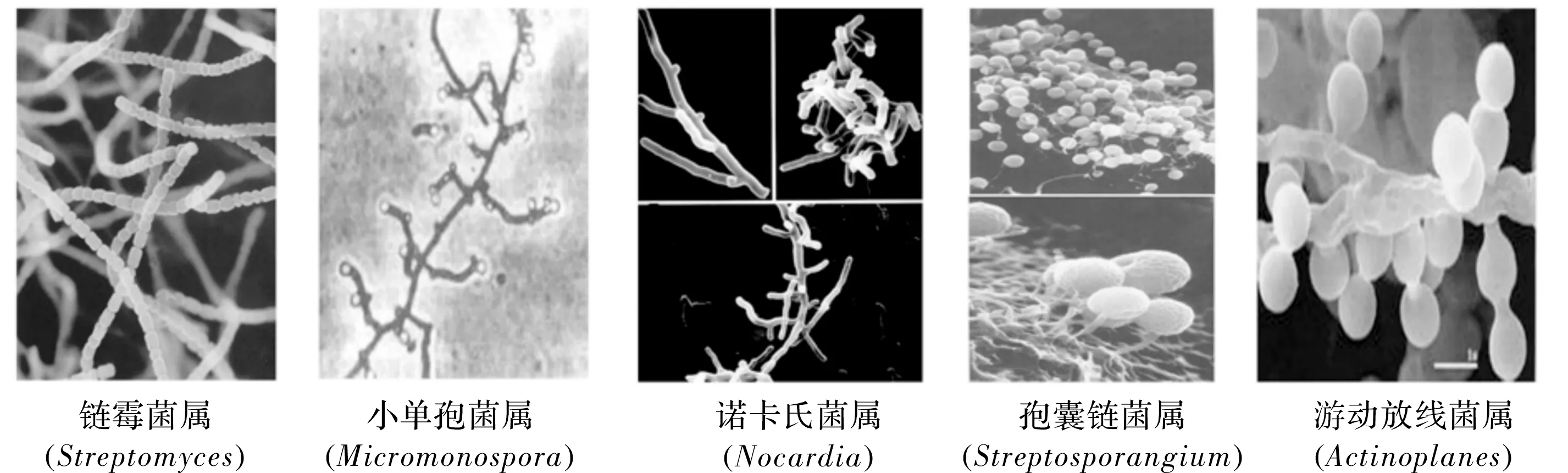
图1 放线菌的代表菌属
2 海洋放线菌的发现和证实
近些年来,日益加剧的病原微生物耐药性以及传统土壤来源材料中已知放线菌和抗生素的大量重复分离,都极大地阻碍了人们寻求新型有效生物活性物质的进程[2].于是,微生物天然产物的分离范围逐步从陆地移向了海洋.占据地球总面积约70%的海洋,无论是从微生物数量还是从多样性上都比陆地生境具有更为广阔的研究前景,因此如何去开发海洋环境中的微生物资源就成了人们空前关注的热点.
但海洋放线菌的种群起源问题,一直以来都备受争议,学者们十分关心是否有真正意义上的海洋专属放线菌存在.一些人认为海洋与陆地直接相连、有着密不可分的联系,陆生放线菌尤其是厚壁菌门释放的抗性孢子可以随河流冲刷入海并在休眠状态下保持多年的繁殖发育潜力,因此他们怀疑从海洋中发现的放线菌均来自陆地,是被不断冲刷而最终随河流汇入大海的[3].另有学者报道,距离陆地越近的海域里海洋放线菌越多而远海中的海洋放线菌则相对较少,这也间接支持了海洋放线菌是起源于陆地的说法[4].
另一方面,也有不少科学家坚持相信海洋中必定有专属的土著放线菌类群存在,他们一直锲而不舍地进行着海洋来源放线菌的分离和研究,以期望早日找到支持自己观点的确凿证据.海洋放线菌的采样最早起始于20世纪60年代末期[5].1984年,Helmke等首次对海生红球菌Rhodococcusmarinonascenssp. nov.进行了系统的分类学形态特征描述[6].随后,Fenical小组在1991年发现了第一个依赖海水生长的特殊放线菌家族“MAR1”(后称之为“Salinispora”),只有在培养基中添加海水或特定钠盐后该菌属菌株才能保持正常的生长[7].这些由传统分类学方法(主要是形态学特征分析)描述过的海洋来源放线菌,在一定程度上预示了海洋专一性放线菌存在的可能性,但由于缺乏基因型和进化关系等分子水平上的有力证据,海洋放线菌的研究在当时仍得不到广泛的认可.直到2002年,即在DNA测序技术逐步成熟时,Fenical小组对“MAR1”菌属进行了16S rRNA序列分析,结果表明该菌属属于小单孢菌科,但在基因型和进化关系上却明显独树一帜,不在相应陆生放线菌的范围之内[8].到2005年,在结合了全面的形态学特征分型和基因组测序分析之后,“MAR1”最终被该小组鉴定为第一个确凿的固有海洋性放线菌属,并正式更名为“Salinispora”[9];这一发现无疑成为海洋放线菌和天然产物研究史上的一座重要里程碑,其深远意义不言而喻.自此,海洋放线菌的研究受到了极大的鼓舞和肯定,越来越多的海洋放线菌被分离和鉴定,为海洋放线菌活性次级代谢产物的开发提供了必要的基础.
3 海洋放线菌的分布和种属
海洋放线菌的分布极其广泛,来源也丰富多样,主要包括上层海水[10]、不同地域的浅水或深水沉积物[11-12]、海绵以及海藻[13]等.目前,已从不同来源的海洋栖息环境中分离到近20个属的固有海洋性放线菌,现归纳整理如表1[14].

表1 海洋放线菌新种属

续表1
4 海洋放线菌的生物活性天然产物
海洋放线菌与陆生放线菌在生理学和种系发育上存在显著差异.它们在特殊海洋生存环境(高盐、高压、低温、低光照和寡营养等)的选择压力下,可调整自身次级代谢途径并积累一些结构和活性都明显不同于陆生放线菌的新颖代谢产物;这些陆生微生物所不具备的潜在优势,使海洋放线菌成为未来新天然药物开发的一大资源宝库[13-14].
目前,人们已经从海洋放线菌发酵培养液中检测到各类结构复杂、活性多样的新颖代谢产物,主要包括聚酮、多肽、杂合聚酮-多肽、大环内酯、萜类、醌类和生物碱等.其中,不少化合物都是潜在的有效抗菌剂和抗癌剂,如salinisporamides[15]、abyssomicins[16]和 proximicins[17]等,这为药物先导化合物的筛选提供了宝贵新来源(表2)[13,18].

表2 海洋放线菌来源的天然产物及其生物活性
5 海洋专性放线菌属Salinispora及其潜在药用天然产物
5.1Salinispora菌属
Salinispora菌属是第一个被报道(1991年)和确证(2005年)的专属海洋放线菌,它的发现为海洋微生物和天然产物研究开辟了一个全新的领域.本属菌株最早于1991年由Fenical小组从巴哈马群岛海岸附近的热带近海沉积物中分离获得(当时命名为“MAR1”),后又从来自大西洋、红海、科特斯海等全球范围内的热带和亚热带近海沉积物中得到稳定分离,这表明该属放线菌具有广泛的世界性分布[6-7].
Salinispora菌属属于小单孢菌科(Micromonosporaceae),但与以往的该科陆生放线菌在表型和基因型上都存在明显差异.2005年,Fenical小组正式鉴定Salinispora为第一个海洋专一性放线菌属,并对其形态学、化学和基因组学特征进行了详细而概括的分类学描述[8].
Salinispora是一类不耐酸、好氧的革兰氏阳性细菌,存在于近海岸线或深至2 000 m的海洋沉积物中,也以共生菌的形式存在于海草和藻类中;具有类似相应陆生放线菌的功能,参与海底有机物质的降解[19].Salinispora能形成不间断的分支菌丝,直径在0.25~0.5 μm之间(见图2 A);其细胞壁肽聚糖中含有N-羟乙酰化的胞壁酸和内消旋-二氨基庚二酸,菌体内不含分枝菌酸,但含饱和脂肪酸;利用的糖源主要为半乳糖、阿拉伯糖和木糖.Salinispora的一般生长条件为:pH 7~12、温度10~30 ℃;它最大的特点是只有向培养基中添加了海水或特定浓度的钠盐时才能维持正常生长.在Salinispora的短孢子梗上,通常长有直径为0.8~3.8 μm的孢子,它们在培养菌落上形成一些灰黑色的区域;这些孢子外观光滑、无法运动,可单个或成簇产生.当培养基中营养足够丰富时,Salinispora形成无气生菌丝的橙色或黑色菌落;营养供给贫瘠时,则形成菌丝体并产生可扩散的橙色、粉红、深褐色或黑色色素[6-7](见图2 B).

图2 Salinispora的各种形态
目前,根据16S rRNA基因的系统进化分析结果,将Salinispora菌属分成3大类菌种:Salinisporaarenicola、Salinisporatropica和Salinisporapacifica.Salinisporaarenicola(以下简写为S.arenicola)存在于关岛、红海、巴哈马群岛、帕劳、科特斯海和美国维尔京群岛等地的热带或亚热带海域沉积物中,它具有十分广泛的世界性分布;最佳生长条件为20~28℃和25%~50%的海水,主要能源物质为L-脯氨酸、L-苏氨酸、L-酪氨酸、熊果苷和D-水杨苷[8,19-20].相比之下,Salinisporatropica(以下简写为S.tropica)的分布范围则较窄,目前只在巴哈马群岛的热带海域沉积物中发现过该菌种;其最佳生长温度为15~28℃,以菊粉和半乳糖作为碳源[8,20].Salinisporapacifica(以下简写为S.pacifica)目前在还处在进一步的分类学研究中,已经从帕劳、红海、关岛、大堡礁和斐济群岛等地的海域中分离出该菌种[19].
5.2Salinispora属放线菌的潜在药用天然产物
Salinispora自2005年被鉴定以来,一直受到科研工作者们的密切关注,现已陆续从其发酵培养液中分离出多种化学结构新颖的生物活性物质(见图3);这些次级代谢产物大多具有抗癌、抗菌、抗病毒或抗炎症活性,是潜在的药物先导化合物来源(见表3)[20].其中,salinosporamide A是目前最引人注目的Salinispora天然产物之一,它作为一种极为有效的蛋白酶抑制剂,已经进入治疗癌症的临床试验阶段[21].
研究表明,Salinispora属放线菌产生的次级代谢产物具有菌种特异性,即S.arenicola、S.tropica和S.pacifica这3个菌种的次级代谢产物化学型是不重叠的;这里,用于区分不同菌种的依据是16S rRNA基因进化关系,而不再是传统的形态学分类标准.这一发现暗示Salinispora的种系型特异代谢产物的产生与其适应特定外界生存环境有关;据此,可利用该属放线菌的种系型和次级代谢产物化学型之间的特异性对应关系,去挖掘和发现更多的新天然活性产物,在一定程度上避免化合物的重复筛选[22].

表3 Salinispora属放线菌的次级代谢产物和生物学活性

图3 Salinispora属放线菌代谢产物的化学结构
6 非核糖体多肽(NRP)
6.1 多肽类天然产物
作为天然产物中排名紧跟聚酮之后的第二大家族,多肽类活性物质一直备受人们关注.根据它在生物体内的合成方式,可将其分为两大类:一类是经核糖体途径产生,例如,硫醚抗生素乳酸链球菌肽和枯草菌素是由核糖体合成前体多肽后经翻译后修饰和蛋白酶解加工而成[23];而另一类活性多肽在合成过程中则完全不依赖于常见的蛋白质核糖体合成系统、占据着制药业中更为重要的地位,统称为非核糖体多肽(NRPs;nonribosomal peptides),例如,谷胱甘肽和细菌细胞壁前体二肽DAla-DAla由酶催化的磷酸化作用组装而来[24],短杆菌酪肽(tyrocidine)由短杆菌酪肽合酶(tyrocidine synthetases)合成.
6.2 非核糖体多肽及其多样性
NRPs是一大类在化学结构和生物学活性上都具有多样性的重要多肽类天然产物(见图4和图5),广泛应用于现代医药业、农业和畜牧业中.与核糖体系统合成产物不同,NRP组分中不仅包含20种基本的蛋白质源氨基酸,还含有大量的稀有氨基酸,如D-氨基酸、α-羟酸、N-/O-甲基氨基酸等;而结构上,NRP分子经常呈完整的大环状或杂环状,侧链还被糖基化、磷酸化、酰基化等作用修饰[25].这些独有特征赋予NRPs复杂多样的结构,进而表现出丰富的生物学活性,可概括如下[25-27]:①抗生素:青霉素(penicillins)前体ACV、万古霉素(vancomycin)、杆菌肽(bacitracin)、短杆菌肽(gramicidin);②铁载体:分枝菌素(mycobactin)、肠菌素(enterobactin);③毒素:丁香霉素(syringomycin)、烟曲霉毒素(gliotoxin)、HC-toxin;④免疫抑制剂:雷帕霉素(rapamycin)、环孢霉素(cyclosporin);⑤生物表面活性剂:surfactin A;⑥抗肿瘤:博莱霉素(bleomycin)以及⑦激素、抗真菌、抗病毒等.

图4 陆地环境来源的NRPs

图5 海洋环境来源的NRPs
7 非核糖体多肽合酶(NRPS)
7.1 非核糖体多肽合酶的结构与功能
非核糖体多肽合酶(NRPSs;nonribosomal peptide synthetases)是一类大的多酶复合物,负责非核糖体多肽的生物合成.对大多数真菌非核糖体多肽合酶而言,合成某种特定多肽产物的NRPS是一个单条多肽链,如1 600 000的环孢霉素合酶[28](cyclosporin synthetases);而大多数原核细菌的非核糖体多肽合酶则由一些相互作用的亚基组成,每个亚基又单独称作NRPS,如短杆菌酪肽合酶[29](tyrocidine synthetases)由3个相互作用的亚基组成,分别称为短杆菌酪肽合酶TyrA、TyrB、TyrC.
NRPSs由一系列被称作“模块(module)”的重复催化单位和结构单位组成,一个完整的NRPS通常包含3~15个模块,每个模块负责完成肽链延伸的一轮循环反应,将单个相应的氨基酸组成单元掺入到非核糖体多肽产物组装线的对应位置上.特别的是,每个模块只有以结合上辅因子P-pant(磷酸泛酰巯基乙胺)后的完整形式存在时,才能发挥出其催化活性;辅因子P-pant是非核糖体多肽合酶被翻译后由4’-磷酸泛酰巯基乙胺转移酶一起添加到所有模块上的.NRPS的每个模块又进一步被划分为几个“结构域(domain)”,它们催化单个生化反应步骤,如活化、共价连接、掺入单体底物的选择性修饰、酰胺键形成和多肽产物释放等(图6).
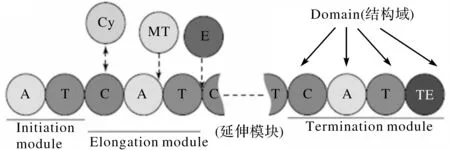
图6 非核糖体多肽合酶的组织结构
一个最小的NRPS链延伸模块包括3个核心的结构域:氨基酸腺苷化结构域(A domain;adenylation domain)、肽酰载体蛋白结构域(PCP domain;peptidyl carrier protein domain;也叫硫酯结构域,T domain)和缩合结构域(C domain;condensation domain).它们催化的一轮链延伸循环反应包括以下3个步骤[25](图7):① 首先,A结构域特异性地结合一个氨基酸底物,在ATP消耗下催化相应“氨酰-AMP”的形成;② 接着,已激活的氨酰-AMP连接到PCP结构域的辅因子Ppant(4’-磷酸泛酰巯基乙胺)的空载巯基上,形成“氨酰-S-PCP”复合物;③ 最后,C结构催化肽链的延伸.分别携带有氨酰基和肽酰基的载体(第一个肽键时是两个带有氨酰基的载体)与C结构域上的特定区域(受体位点a & 供体位点d)结合,氨酰-S-载体复合物上的氨基向肽酰-S-载体复合物上肽酰基的酰基进行亲核攻击,形成新肽键,进而产生延长了一个氨基酸长度的新的肽酰-S-载体复合物和游离的载体[30].
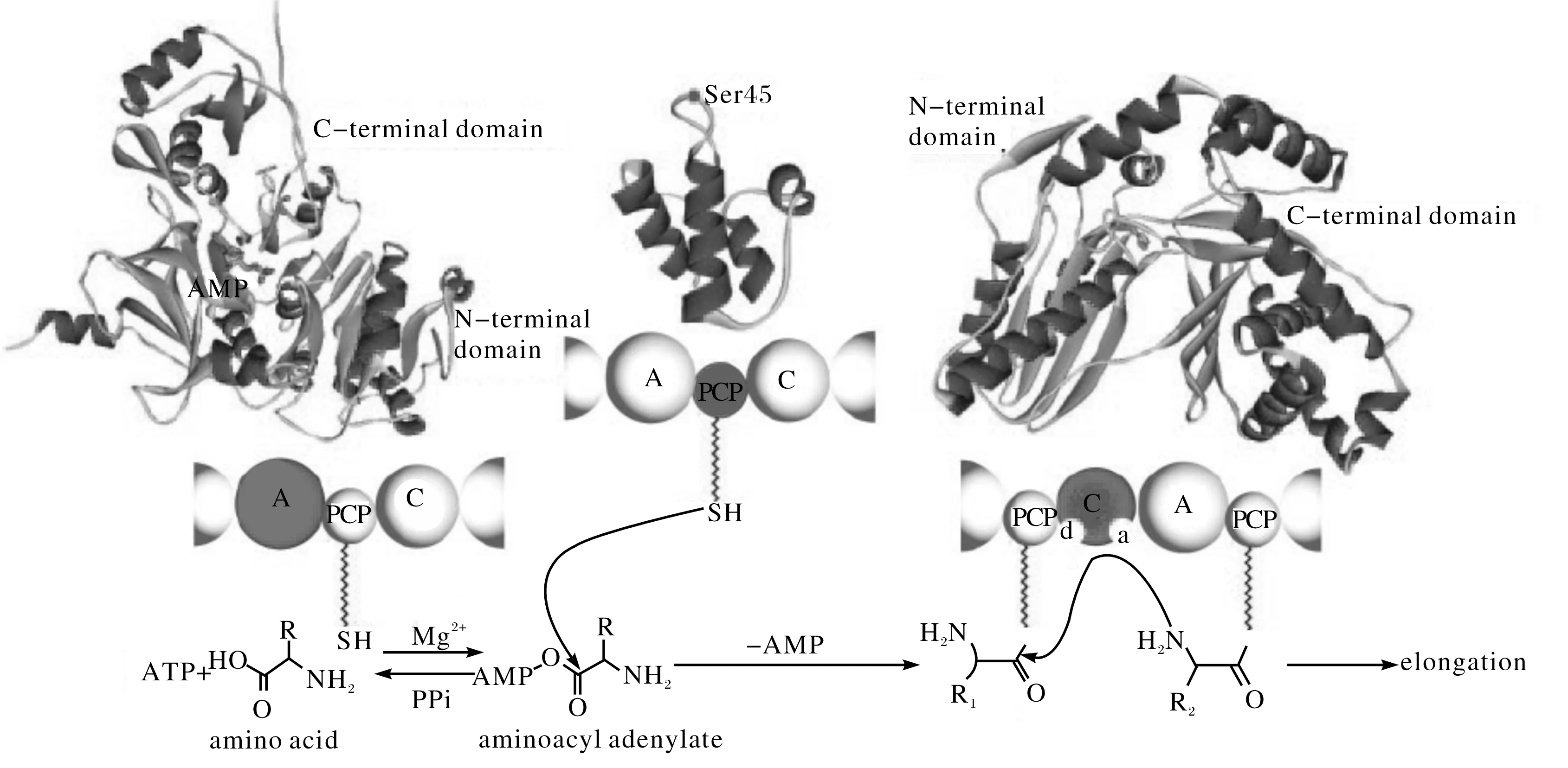
图7 肽链延伸的一轮循环反应(由单个NRPS模块完成)
7.2 非核糖体多肽合酶的类型和研究意义
根据NRP组装时NRPSs所采用的不同生物合成策略,可将其分为3类:线性NRPSs(A型)、迭代型NRPSs(B型)、非线性NRPSs(C型).线性NRPSs(A型)中,3个核心结构域以C-A-PCP的顺序在延伸模块上排列,即一个合成含n个氨基酸残基的多肽的线性NRPS蛋白质模板由结构域以A-PCP-(C-A-PCP)n-1-Te顺序排列的n个模块组成.另一方面,NRPs的合成方向是从N端到C端,故A型NRPS模块的数量、种类、排列顺序决定了多肽产物的一级结构,二者成线性关系.与线性NRPSs(A型)相比,迭代型NRPSs(B型)的特点是在多肽合成过程中多次使用它们的模块或结构域;而非线性NRPSs(C型)的核心结构域C、A、T中至少有一个异常排列,其模块数与多肽产物的氨基酸残基数无线性对应关系[31].
显然,NRPSs的模块构造特征为发展NRP组合生物合成技术提供了一个有力的机会,我们可以通过人工操控NRP生物合成途径中的特定基因(如结构域或模块的互换)来预测和生产具有新的生物学活性的杂合多肽,为抗生素新药研究和开发奠定良好基础.
8 腺苷化酶超家族与NRPS氨基酸腺苷化结构域
8.1 腺苷化酶超家族
腺苷化酶超家族(adenylating enzyme superfamily)是指一类在ATP消耗下将特定羧酸底物激活而生成相应“酰基-AMP”的酶类的总称,它们在原核生物和真核生物的代谢途径中均发挥极其重要而多样化的作用,参与核糖体途径和非核糖体途径多肽合成、脂肪酸氧化和合成、酶类调节等一系列代谢过程.这类酶都能催化以下两步特征性反应:①结合特异性羧酸底物,与ATP缩合形成有反应活性的腺苷化底物并生成焦磷酸(PPi);②活化的腺苷化底物与亲核试剂反应,释放出终产物和AMP.
根据氨基酸序列同源性和识别底物特异性,将腺苷化酶超家族分成3大类:NAL家族、氨酰-tRNA合酶和NIS(非NRPS依赖的铁载体合酶).其中,NAL家族又由酰基/芳基辅酶A合成酶、荧光素氧化酶和NRPS氨基酸腺苷化结构域(NRPS A domain)组成[32].NAL家族的最大特点是其成员在空间结构上具有高度一致的同源性:他们都折叠成一个较大的N-末端结构域和一个较小的C-末端结构域,中间则由一段柔性铰链连接;而酶的活性中心通常正位于N-、C-这两个相邻折叠结构域的交界处,因此酶结合底物后N-、C-两个折叠结构域之间的相对空间定向也会随之改变[33-35].目前,已有许多腺苷化酶家族成员的晶体结构得到解析,如萤火虫荧光素酶[34]、短杆菌肽合酶的PheA[35]、Bacillibactin合酶的DhbE[36]、乙酰辅酶A合酶[37]、4’-叶绿素苯甲酸酯:CoA合酶[38]等.
8.2 NRPS氨基酸腺苷化结构域(NRPS A domain)
NRPS氨基酸腺苷化结构域(NRPS A domain,简称A结构域)是“腺苷化酶超家族”的一员,平均大小约为550个氨基酸残基,在NRP生物合成途径中起两方面的作用:①选择和激活特异性氨基酸底物,如图8(A)所示;②转移活化的氨酰-AMP到下游T结构域的Ppant上,如图8(B)所示.由此可见,NRPS A domain在NRP生物合成中占据着重要的地位,它决定了NRP终产物的成分组成和结构多样性.
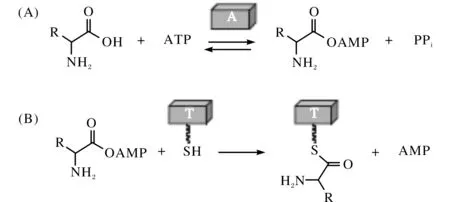
图8 NRPS A domain催化的反应
A结构域的底物特异性与空间结构之间的关联,一直是相关领域科学家们的研究热点.第一个得到晶体结构解析的A结构域 —— 短杆菌肽合酶(gramicidin synthetase)中的PheA,于1997年由德国科学家Mohamed A Marahiel带领的团队率先完成[35](图9).基于此晶体结构学研究成果,Mohamed A Marahiel等科学家将PheA和DhbE活性位点处的氨基酸残基序列与其他底物特异性已知的NRPS A结构域的对应位置上的氨基酸残基序列进行比对分析和进化树分析,确定了由底物结合口袋中的10个氨基酸残基依次排列组成的序列标签,他们将其称作非核糖体多肽生物合成的“底物特异性赋予的密码子”[39].在核糖体合成体系中,3个碱基决定1个核糖体多肽合成的密码子,而NRPS合成体系中则是由其NRPS A结构域序列标签的10个氨基酸残基的序列特性来决定1个NRP密码子.因此,只需通过生物信息学方法分析NRPS A结构域的一级氨基酸序列,就可以使用以上NRP密码子规律,在酶活性测定之前预测一些未知NRPS A结构域的特异性底物.Challis等将已通过实验和其它途径确定的NRPS A结构域有关信息收集整理起来,建立了其氨基酸底物识别特异性的数据库,可通过相关网址进行查寻(http://raynam.chm.jhu.edu),这极大地方便了NRPS A结构域底物特异性的研究和预测[40].
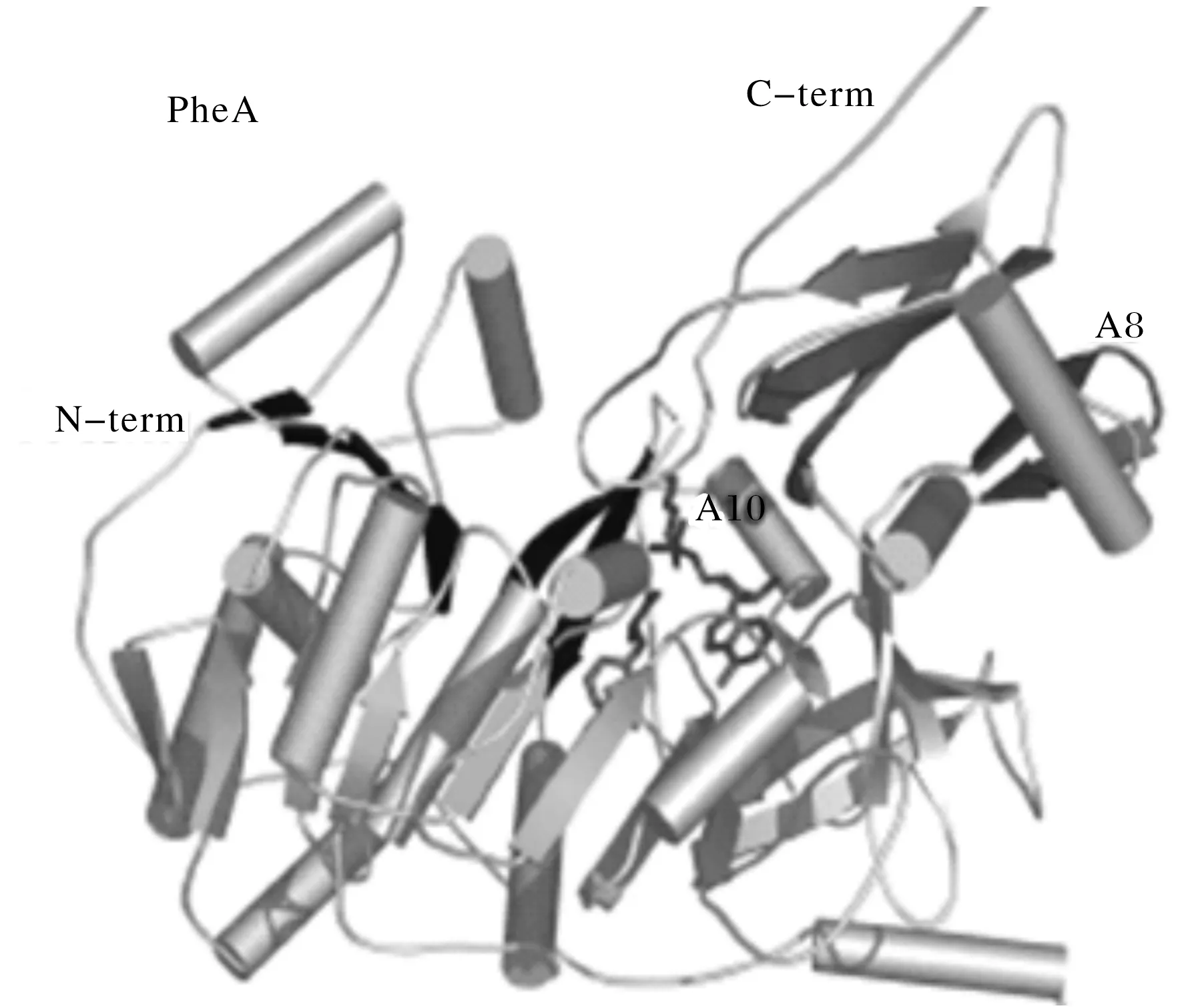
图9 短杆菌肽合酶中PheA的晶体结构示意图
通常,A结构域具有高度的底物特异性;然而也发现有一些底物特异性较宽松的天然A结构域存在[41],它们在组合生物合成上具有重大利用价值,是创造非天然合成途径的潜在试剂和工具,能用于产生新的药物先导化合物.然而,分析A结构域酶活性的传统经典方法是测定具有放射性的ATP-[32P]PPi交换反应的酶动力学常数.这种方法不利于从大批量测试对象中筛选出个别具有较低底物特异性的A结构域,因为世界上大部分实验室都不具备处理高通量放射性物质的能力.2009年Thomas J. McQuade等报到了一种便捷而非放射性的高通量确定A结构域酶动力学性质的比色测定法[42].他们在A结构域催化氨酰-AMP形成的反应体系中添加无机焦磷酸酶和孔雀石绿染料,这样由A结构域作用于ATP和底物后释放出的焦磷酸就会分解成正磷酸Pi,孔雀石绿染料能和产生正磷酸Pi结合并在620 nm处有最大光吸收,故最后只需测定正磷酸的浓度即反应产物的OD620值就可获得A结构域的各项酶学参数.该比色测定法准确可靠而定量,可在96或384孔板中进行.这项工作为大批量筛选和鉴定A结构域突变体提供了一个有力的工具,极大地推动了A结构域在NRP组合生物合成学上的应用和新NRP先导化合物的探索研究.
我们课题组正在开展海洋放线菌的相关研究,在cDNA文库构建、调控因子、A结构域取得了一些初步进展[43-48].
[1] Berdy J. Bioactive microbial metabolites[J]. J Antibiot, 2005, 58(1):1-26.
[2] Newman D J, Cragg G M, Snader K M. Natural products as sources of new drugs over the period 1981-2002[J]. J Nat Prod, 2003, 66(7):1022-1037.
[3] Goodfellow M, Haynes J A. Actinomycetes in Marine Sediments[M]. New York: Academic Press Inc, 1984: 453-472.
[4] Maldonado L A, Stach J E, Pathomaree W, et al. Diversity of cultivable actinobacteria in geographically widespread marine sediments[J]. Antonie Van Leeuwenhoek, 2005, 87(1):11-18.
[5] Weyland H. Actinomycetes in North Sea and Atlantic Ocean sediments[J]. Nature, 1969, 223(5208):858.
[6] Helmke E, Weyland H.Rhodococcusmarinonascenssp. nov., an actinomycete from the sea[J]. Int J Syst Bacteriol, 1984, 34(2):127-138.
[7] Jensen P R, Dwight R, Fenical W. Distribution of actinomycetes in near-shore tropical marine sediments[J]. Appl Environ Microbiol, 1991, 57(4):1102-1108.
[8] Mincer T J, Jensen P R, Kauffman C A, et al. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments[J]. Appl Environ Microbiol, 2002, 68(10):5005-5011.
[9] Maldonado L A, Fenical W, Jensen P R, et al.Salinisporaarenicolagen.nov.,sp. nov. andSalinisporatropicasp. nov., obligate marine actinomycetes belonging to the family Micromonosporaceae[J]. Int J Syst Evol Microbiol, 2005, 55(5):1759-1766.
[10] Gross H. Strategies to unravel the function of orphan biosynthesis pathways: recent examples and future prospects[J]. Appl Microbiol Biotechnol, 2007, 75(2):267-277.
[11] Bowers A A, Acker M G, Koglin A, et al. Manipulation of thiocillin variants by prepeptide gene replacement: structure, conformation, and activity of heterocycle substitution mutants[J]. J Am Chem Soc, 2010, 132(21):7519-7527.
[12] Pathomaree W, Stach J E, Ward A C, et al. Diversity of actinomycetes isolated from challenger deep sediment (10,898 m) from the Mariana Trench[J]. Extremophiles, 2006, 10(3):181-189.
[13] Gulder T A, Moore B S. Chasing the treasures of the sea-bacterial marine natural products[J]. Curr Opin Microbiol, 2009, 12(3):252-260.
[14] Ward A C, Bora N. Diversity and biogeography of marine actinobacteria[J]. Curr Opin Microbiol, 2006, 9(3):279-286.
[15] Lam K S. Discovery of novel metabolites from marine actinomycetes[J]. Curr Opin Microbiol, 2006, 9(3):245-251.
[16] Fenical W, Jensen P R. Developing a new resource for drug discovery: marine actinomycete bacteria[J]. Nat Chem Biol, 2006, 2(12):666-673.
[17] Reed K A, Manam R R, Mitchell S S, et al. Salinosporamides D-J from the marine actinomyceteSalinisporatropica, bromosalinosporamide, and thioester derivatives are potent inhibitors of the 20S proteasome[J]. J Nat Prod, 2007, 70(2):269-276.
[18] Riedlinger J, Reicke A, Zahner H, et al. Abyssomicins, inhibitors of the paraaminobenzoic acid pathway produced by the marineVerrucosisporastrain AB-18-032[J]. J Antibiot, 2004, 57(4):271-279.
[19] Fiedler H P, Bruntner C, Riedlinger J, et al. Proximicin A, B and C, novel aminofuran antibiotic and anticancer compounds isolated from marine strains of the actinomyceteVerrucosispora[J]. J Antibiot, 2008, 61(3):158-163.
[20] Zotchev S B. Marine actinomycetes as an emerging resource for the drug development pipelines[J]. J Biotechnol, 2012, 158(4):168-175.
[21] Jensen P R, Gontang E, Mafnas C, et al. Culturable marine actinomycete diversity from tropical Pacific Ocean sediments[J]. Environ Microbiol, 2005, 7(7):1039-1048.
[22] Jensen P R, Mafnas C. Biography of the marine actinomyceteSalinispora[J]. Environ Microbiol, 2006, 8(11):1881-1888.
[23] Jensen P R, Williams P G, Oh D C, et al. Species-specific secondary metabolite production in marine Actinomycetes of the genusSalinispora[J]. Appl Environ Microbiol, 2007, 73(4):1146-1152.
[24] Feling R H, Buchanan G O, Mincer T J, et al. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genusSalinispora[J]. Angew Chem Int Ed Engl, 2003, 42(3):355-357.
[25] Penn K, Jenkins C, Nett M, et al. Genomic islands link secondary metabolism to functional adaptation in marine Actinobacteria[J]. ISME J, 2009, 3(10):1193-1203.
[26] Chatterjee C, Paul M, Xie L, et al. Biosynthesis and mode of action of lantibiotics[J]. Chem Rev, 2005, 105(2):633-684.
[27] Fan C, Park I S, Walsh C T, et al. D-alanine ligase: phosphonate and phosphinate intermediates with wild type and the Y216F mutant[J]. Biochemistry, 1997, 36(9):2531-2538.
[28] Grunewald J, Marahiel M A. Chemoenzymatic and template-directed synthesis of bioactive macrocyclic peptides[J]. Microbiol Mol Biol Rev, 2006, 70(1):121-146.
[29] Sieber S A, Marahiel M A. Learning from nature’s drug factories: nonribosomal synthesis of macrocyclic peptides[J]. J Bacteriol, 2003, 185(24):7036-7043.
[30] Challis G L, Naismith J H. Structural aspects of nonribosomal peptide biosynthesis[J]. Curr Opin Struct Biol, 2004, 14(6):748-756.
[31] Weber G, Schorgendorfer K, Schneider-Scherzer E, et al. The peptide synthetase catalyzing cyclosporine production in Tolypocladium niveum is encoded by a giant 45.8-kilobase open reading frame[J]. Curr Genet, 1994, 26(2):120-125.
[32] Mootz H D, Marahiel M A. The tyrocidine biosynthesis operon of Bacillus brevis: complete nucleotide sequence and biochemical characterization of functional internal adenylation domains[J]. J Bacteriol, 1997, 179(21):6843-6850.
[33] Mootz H D, Marahiel M A. Biosynthetic systems for nonribosomal peptide antibiotic assembly[J]. Curr Opin Chem Biol, 1997, 1(4):543-551.
[34] Mootz H D, Schwarzer D, Marahiel M A. Ways of assembling complex natural products on modular monribosomal peptide synthetases[J]. Chembiochem, 2002, 3(6):490-504.
[35] Schmelz S, Naismith J H. Adenylate-forming enzymes[J]. Curr Opin Struct Biol, 2009, 19(6):666-671.
[36] Gulick A M. Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-Ribosomal peptide synthetases, and firefly luciferase[J]. ACS Chem Biol, 2009, 4(10):811-827.
[37] Conti E, Franks N P, Brick P. Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes[J]. Structure, 1996, 4(3):287-298.
[38] Conti E, Stachelhaus T, Marahiel M A, et al. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S[J]. EMBO J, 1997, 16(14):4174-4183.
[39] May J J, Kessler N, Marahiel M A, et al. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases[J]. Proc Natl Acad Sci USA, 2002, 99(19):12120-12125.
[40] Gulick A M, Starai V J, Horswill A R, et al. The 1.75 Å crystal structure of acetyl-CoA synthetase bound to adenosine-5’-propylphosphate and coenzyme A[J]. Biochemistry, 2003, 42(10):2866-2873.
[41] Gulick A M, Lu X, Dunaway-Mariano D. Crystal Structure of 4-Chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states[J]. Biochemistry, 2004, 43(27):8670-8679.
[42] Stachelhaus T, Mootz H D, Marahiel M A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases[J]. Chem Biol, 1999, 6(8):493-505.
[43] 马艳玲, 邓 海, 刘中来, 等. 稀有海洋放线菌Salinisporaarenicola大片段DNA基因组文库的构建[J].生物技术, 2010(3):1-3.
[44] 马艳玲, 邓 海, 刘中来, 等. 海洋放线菌Streptomycessp.大片段DNA基因组文库的构建[J].生物技术, 2010(5):1-3.
[45] 马艳玲, 邓 海, 魏菁菁, 等.稀有海洋放线菌Salinisporaarenicola非核糖体肽合成酶和卤代酶生物合成基因簇核心区的克隆及序列分析[J].生物技术通报, 2011(2):157-162.
[46] Sisi Xia, Yanlin Ma, Wei Zhang, et al. Identification of Sare0718 as an alanine-activating adenylation domain in marine actinomyceteSalinisporaarenicolaCNS-205[J]. PLoS ONE, 2012, 7(5): e37487.
[47] Xiao Shu, Wei Zhang, Jinlong Yu, et al. Possible Negative Regulatory Effects of Antibiotic Production in a Hetrologous System by Sare_4854, a Novel Member ofStreptomycesAntibiotic Regulatory Protein Family (SARP) from a Marine MicroorganismSalinisporaarenicolaCNH643 DSM 44819[J]. Journal of Pure and Applied Microbiology, 2014,8(2):1447-1452.
[48] 陆胜利, 祁 超. 海洋放线菌对S.areniocolaCNS-205腺苷化结构域基因的克隆、表达和纯化[J].华中师范大学学报:自然科学版, 2014, 48(6):876-884.
Progress of Metabolites,NRP and adenylation domain from marine actinomycete
LU Shengli1, QI Chao2
(1.Department of Medical, Anqing Medical College, Anqing, Anhui 246052;2.Hubei Key Laboratory of Genetic Regulation and Integrative Biology, College of Life Sciences,Central China Normal University, Wuhan 430079)
Metabolites nonribosomal peptide synthetases and Adenylation domain of Marine actinomycete were summarized.
marine actinomycete; metabolites; adenylation domain
2014-01-20.
国家自然科学基金项目(20772040);教育部中央高校自主研究项目(CCNU14F01006).
1000-1190(2015)01-0114-11
Q936
A
*通讯联系人. E-mail: qichao@mail.ccnu.edu.cn.
