催化动力学荧光法测定头发中痕量钼
2015-03-20宋少飞周福林
宋少飞,李 平,周福林
(1.运城学院应用化学系,山西 运城044000;2.集宁师范学院化学系,内蒙古 集宁012400)
钼作为一种微量元素,是生命体必需的,它参与人体内多种酶的组成及酶催化作用,在抗癌、心血管保护、细胞内电子传递、阻止龋齿和结石等方面具有非常重要的作用. 钼元素缺乏会导致肾结石、心血管病、食道癌等多种病症的发生;而过量的钼则损伤生殖细胞,干扰人体正常的性机能和身体发育[1].
目前,相关研究表明,原子吸收法[2-4]、分光光度法[5-8]、荧光光度法[9-11]、色谱法[12]、极谱法[13]以及催化光度法[14-16]等已用于样品中钼含量的测定.其中,催化动力学荧光分析法具有灵敏度高、选择性好等优点,近年来有关此法测定钼的报道也不少[17-20].但上述方法大多反应体系较复杂,而且有的用到组分较多的表面活性剂[19-20],因而给配制溶液、实验操作带来不便.本文研究发现,在酸性体系中,钼(VI)离子的存在,可以大大催化加速溴酸钾与二氯荧光素的氧化褪色反应.基于此实验事实,一种用于测定痕量钼(VI)的催化动力学荧光法成功建立.相关实验数据表明,该方法具有较高的灵敏度,较强的选择性,而且操作简便,对人发中钼(VI)含量的测定结果良好.
1 实验部分
1.1 主要试剂及仪器
仪器:PerkinElmer LS55 型荧光分光光度计(美国PE 公司);BSA124S 电子分析天平(长沙市秋龙仪器设备有限公司);HHS21-Ni8 型恒温水浴锅(北京三二八科学仪器有限公司);烘箱(天津市华北实验仪器有限公司);202-18.DB-1A 电热板(南京庚辰科学仪器有限公司).
试剂:钼标准储备液1 mg/mL;配制方法:精确称取0.9208g 钼酸铵于400 mL 烧杯中,用二次蒸馏水定容到500 mL 容量瓶中;KBrO3:0.1 mol/L;H2S04:1 mol/L;二氯荧光素:1 ×10-3mol/L;NaOH 溶液:6 mol/L;混酸∶ 硝酸∶ 高氯酸为5∶ 1 溶液;试剂均为分析纯,实验用水经二次蒸馏处理.
1.2 样品的采集与实验方法
1.2.1 发样的采集方式
对20 名志愿者进行采样,样本的构成不考虑研究对象的年龄和性别.使用不锈钢剪刀,剪取枕部离发根1 ~2 cm 处2 ~3 g 头发,分别装入专用塑料袋中.共采集头发样品20 件,分为5 组.
1.2.2 实验方法
向一支50 mL 比色管中加入一定量的钼(VI)标准溶液,再分别加入5mL 0.1mol/L 的KBrO3溶液,1.5mL1mol/L 的H2SO4溶液,以及3.5mL 1 ×10-3mol/L 的二氯荧光素溶液,添加完毕后用水定容,摇匀待用.另取一支50mL 比色管,在不加入钼(VI)标准溶液的情况下,按上述方法制备一份空白溶液,以作对比.然后,将两支比色管置于80℃的热水中,水浴加热10min,取出后用流水冷至室温.以272nm 为激发波长,547nm 为发射波长调整荧光光度计,分别测定比色管中溶液的荧光强度,记录数据.其中,非催化反应荧光强度为I0,催化反应荧光强度为I,计算强度差:ΔIF= I0-I.
2 实验结果与讨论
2.1 反应体系荧光行为
为了确定Mo(VI)对KBrO3氧化二氯荧光素褪色反应的催化活性,配制不同体系的溶液,用荧光分光光度计分别测定各反应体系的荧光行为,给出各体系的激发和发射光谱(见图1).从图中可以看出,与曲线1,1′ 相比,曲线2,2′ 强度大幅度下降,表明H2SO4溶液中,KBrO3可以缓慢氧化二氯荧光素褪色.向二氯荧光素与KBrO3的混合体系中加入Mo(VI)后,相同时间内,其激发与发射曲线强度明显降低,表明褪色反应速率加快,确定了Mo(VI)的催化活性(曲线3、3′).但需注意的是,添加Mo(VI)前后,光谱形状没有改变,说明Mo(VI)在反应体系中仅起催化作用而不会影响反应产物的结构.据此建立了催化动力学荧光法测定痕量钼的方法.
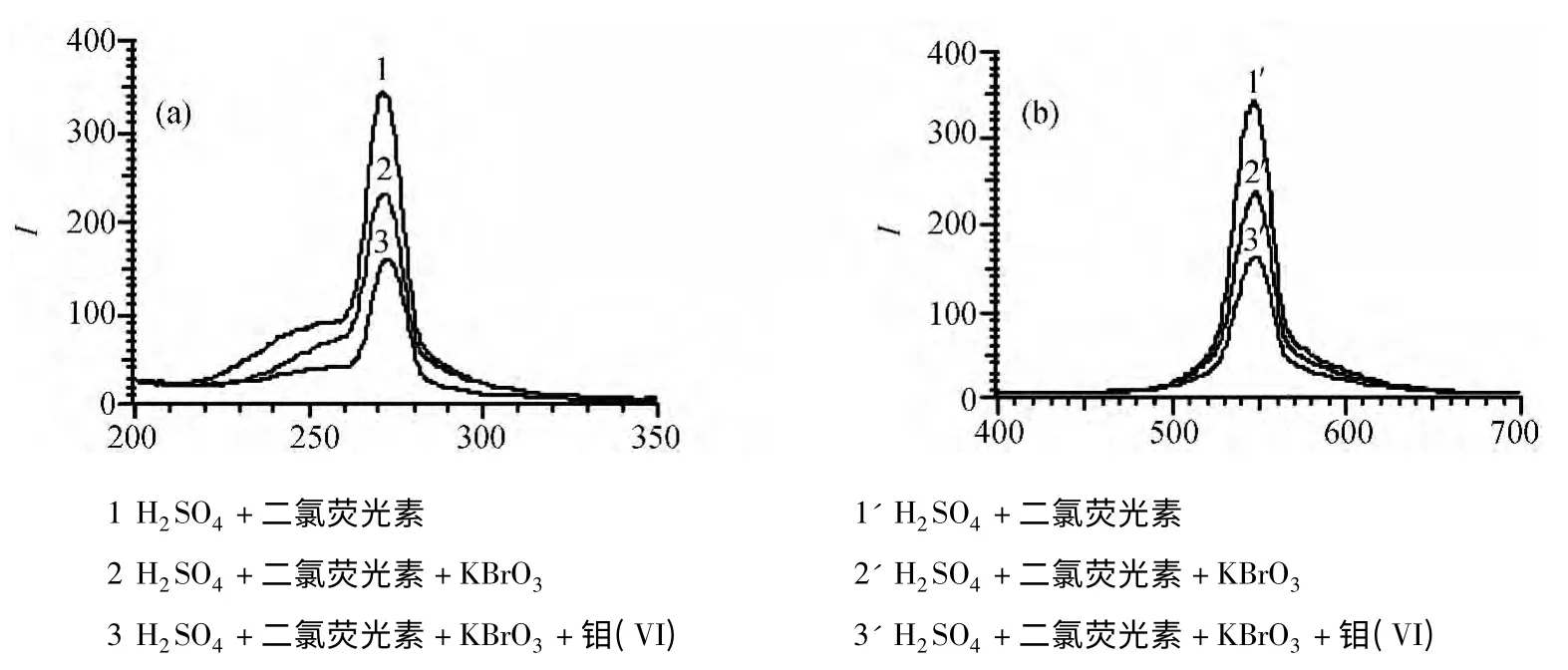
图1 反应体系荧光光谱行为:(a)激发与(b)发射Fig. 1 Fluorescence spectrum:(a)Excitation and (b)emission
2.2 实验条件的优化
2.2.1 最大激发和发射波长的确定
不同反应体系的荧光光谱行为如图1 所示,从中可以看出,加入氧化剂KBrO3与催化剂Mo(VI)前后,各体系的最大激发与发射波长均分别位于272 nm 和547 nm 处.基于以上各体系荧光光谱行为,本实验确定最大激发和发射波长分别为272 nm 和547 nm.
2.2.2 氧化底物浓度的影响
本实验以二氯荧光素为氧化底物,因此,首先考察了二氯荧光素溶液浓度对体系荧光强度的影响(见图2).由图可见,在1×10-5~1×10-4mol/L 范围内,随着二氯荧光素浓度的增加,ΔIF值亦增大,但是底物浓度大于7×10-5mol/L 时,ΔIF值随着浓度的增加反而减小.曲线的变化趋势表明,过多的二氯荧光素不利于褪色反应,二氯荧光素溶液浓度为7×10-5mol/L 时体系荧光强度最大,为本实验最佳的底物浓度.
2.2.3 反应介质酸性的影响
本实验在酸性介质硫酸溶液中进行,考察了其浓度对体系荧光强度的影响(见图3).图中曲线变化表明,浓度为3 ×10-2mol/L 时,体系ΔIF最大.因此,确定本实验最佳硫酸溶液浓度为3 ×10-2mol/L.
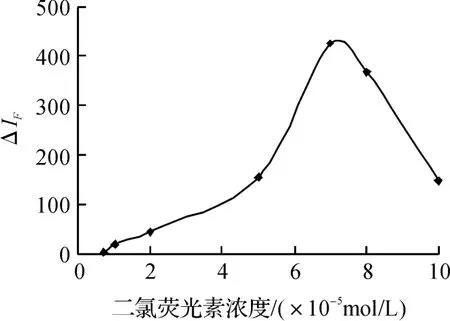
图2 二氯荧光素浓度的影响Fig. 2 Effect of concentration of Dichloro-fluorescein

图3 硫酸溶液浓度的影响Fig.3 Effect of concentration of H2SO4
2.2.4 氧化剂浓度的影响
KBrO3作为氧化剂,其浓度必然对体系荧光强度具有重要的影响,在1 ×10-3~3 ×10-2mol/L 范围内考察了KBrO3浓度与ΔIF值的对应关系(见图4).图中曲线变化行为表明,初始,ΔIF值随着KBrO3溶液增加而增大,当浓度达到1 ×10-2mol/L 时,ΔIF最大.然后,ΔIF值随着KBrO3溶液增加而降低.因此,本实验确定最佳的KBrO3溶液浓度为1 ×10-2mol/L.
2.2.5 体系温度的影响
反应体系温度对氧化剂及催化剂活性均具有重要影响,一般而言,随着温度升高,催化剂活性增加.但过高的温度会导致催化剂失活[21].在实验确定了底物、介质酸性、氧化剂浓度的情况下,分别在不同温度下测定了ΔIF值,考察了温度对荧光强度的影响(见图5).从图中可以看出,反应体系温度为80 ℃时,ΔIF达到最大值.所以本实验均在80 ℃水浴中进行.
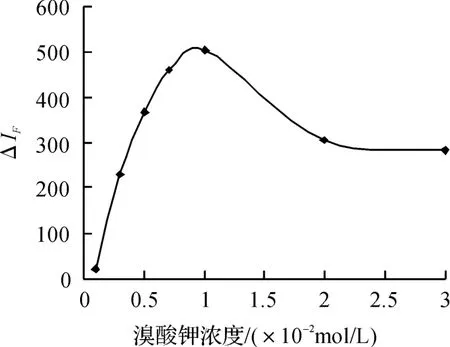
图4 溴酸钾浓度的影响Fig.4 Effect of concentration of KBrO3

图5 体系温度的影响Fig. 5 Effect of reaction temperature
2.2.6 反应时间的影响
在上述最佳条件下,考察了反应时间对体系荧光强度的影响(见图6). 由图可见,当反应时间为10 min 时,ΔIF最大.所以,确定本实验的最佳反应时间为10 min.
2.3 标准曲线
按实验方法,在最佳条件下,对不同浓度的Mo(Ⅵ)标准溶液进行荧光强度测定.分别以Mo(Ⅵ)标准溶液浓度与荧光强度为参数制作标准曲线(见图7),可以看出,在7 ×10-7~1 ×10-5mg/mL 范围内,Mo(Ⅵ)浓度与ΔIF具有良好的线性关系.拟合后得回归方程为:ΔIF=2.178c+32.72(c:×10-6mg/mL),r=0.9918.
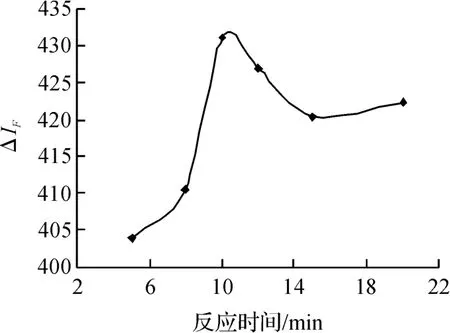
图6 反应时间的影响Fig. 6 Effect of reaction time

图7 标准曲线Fig. 7 Calibration graph
2.4 精密度和检出限
精密度是衡量反应体系稳定性与否的一个重要指标,对浓度为1×10-6mg/mL 的Mo(Ⅵ)标准溶液进行了11 次平行测定,计算出相对标准偏差为0.6%,表明本文建立的催化荧光法体系稳定,系统误差较小.另外,根据IUPAC 建议,计算所得Mo(Ⅵ)的测定检出限为9×10-8mg/mL,显示了该体系较高的灵敏度.
2.5 共存离子干扰实验
进行了共存无机离子对Mo(Ⅵ)(1 ×10-6mg/mL)荧光行为的干扰试验,实验结果表明,当相对误差在±5%时,共存离子的允许倍数为:(800 倍);Zn2+(1500 倍);Ca2+、Hg2+、、Al3+(6.5 倍);Pb2+(5倍);Co2+、Cd2+、、Mn2+、、Cr3+(2 倍);Fe3+、、Sn4+(1 倍).在对实际样品人发测定中发现,干扰离子超过允许倍数造成干扰时,可以使用EDTA 进行掩蔽.
2.6 样品分析
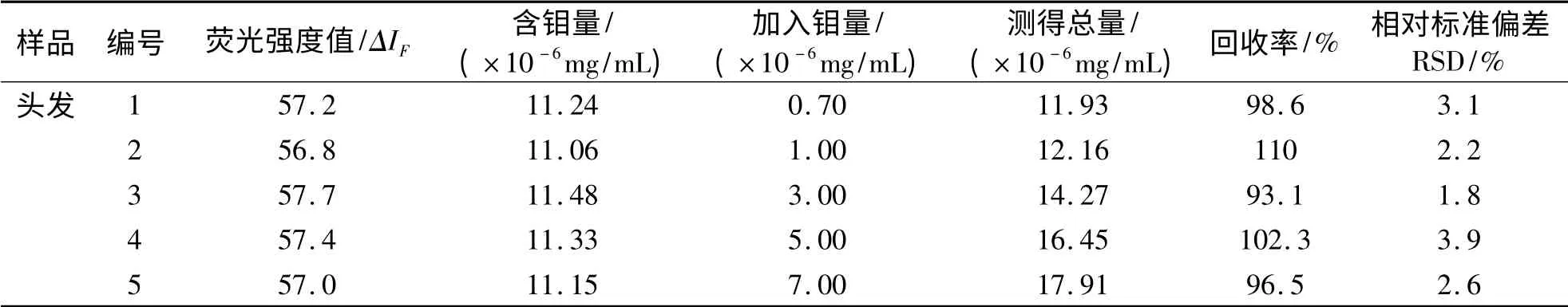
表1 头发样品中钼(VI)含量测定的实验数据Tab.1 Data on determination of Mo(VI)in hair and recovery
采集一定量的人发试样,置于小烧杯中用丙酮预洗,再用中性洗涤剂洗涤,然后用热的蒸馏水冲洗干净,置于60 ℃的烘箱内,用不生锈的剪刀剪成2 cm 左右的小段,待用.准确称取0.4000 g 发样于250 mL的烧杯中,在通风条件下,加入硝酸-高氯酸混合酸(5∶ 1)10 mL.在80 ~100 ℃的水浴中加热溶解,然后转移到电热板上加热,溶液由棕红色转为浅黄色并蒸至近干,浓烟消失,冷却后加少量水(若有颜色用活性炭脱色除去),然后定容于100 mL 容量瓶中,待用.准确移取待测液1 mL,按实验方法进行测定,实验测得结果列于表1.
3 结 论
基于钼(VI)对溴酸钾氧化二氯荧光素褪色反应的催化活性,成功建立了可用于测定痕量钼(VI)的催化荧光法,与其它已有方法相比,催化荧光法灵敏度高,选择性强,而且实验操作较为简便.方法的线性范围为7 ×10-7~1 ×10-5mg/mL,检出限为9 ×10-8mg/mL,对浓度为1 ×10-6mg/mL 的钼(VI)分别平行测定11 次,得相对标准偏差(RSD)为0.6%.已用于人发中痕量钼(VI)的测定,结果令人满意.
[1]吴茂江.钼与人体健康[J].微量元素与健康研究,2006,23(5):66-67.
[2]Filik H,Çengel T,Apak R. Selective cloud point extraction and graphite furnace atomic absorption spectrometric determination of molybdenum(VI)ion in seawater samples[J]. Journal of Hazardous Materials,2009,169(1-3):766-771.
[3]Gürkan R,Aksoy Ü,Ulusoy H. Determination of low levels of molybdenum(VI)in food samples and beverages by cloud point extraction coupled with flame atomic absorption spectrometry[J]. Journal of Food Composition and Analysis,2013,32(1):74-82.
[4]夏行昊,姜毓君,单艺.石墨炉原子吸收光谱法测定婴幼儿食品及乳品中钼[J].中国乳品工业,2014,42(5):58-59.
[5]车音,陈松庆,朱霞石.浊点萃取分离-光度法测定水中痕量钼[J].理化检验-化学分册,2010,46(6):618-620.
[6]王君玲,王科伟,樊月琴,等.新试剂联苯基双荧光酮分光光度法测定微量钼[J].光谱实验室,2013,30(4):1836-1839.
[7]Gharehbaghi M,Shemirani F. Ionic liquid-based dispersive liquid-liquid microextraction and enhanced spectrophotometric determination of molybdenum(VI)in water and plant leaves samples by FO-LADS[J]. Food and Chemical Toxicology,2011,49(2):423-428.
[8]Lešková M,Sklenárˇová H,Bazel Y,et al. A non-extractive sequential injection method for determination of molybdenum[J]. Talanta,2012,96(15):185-189.
[9]Jiang C Q,Wang J Z,He F. Spectrofluorimetric determination of trace amounts of molybdenum in pig liver and mussels[J]. Analytica Chimica Acta,2001,439(2):307-313.
[10]尚虹.荧光光度法测定自来水及尿样中痕量钼[J].中国卫生检验杂志,2009,19(7):1505-1506.
[11]乔善宝,王庆东,焦昌梅.异丙肾上腺素荧光分光光度法测定豆样中痕量钼[J].理化检验-化学分册,2009,45(9):1051-1052;1055.
[12]刘德晔,谷静,刘华良.离子色谱-电感耦合等离子体质谱联用研究尿中碘和钼形态[J].分析实验室,2013,32(7):40-44.
[13]付爱瑞,肖凡,罗治定,等.氢氧化锰共沉淀分离-催化极谱法测定土壤中有效钼[J].理化检验-化学分册,2012(4):417-419.
[14]樊雪梅,崔孝炜.催化动力学光度法测定钼尾矿中痕量钼(Ⅵ)[J].分析科学学报,2014,30(1):127-129.
[15]Nakano S,Kamaguchi C,Hirakawa N. Flow-injection catalytic spectrophotometic determination of molybdenum(VI)in plants using bromate oxidative coupling of p-hydrazinobensenesulfonic acid with N-(1-naphthyl)ethylenediamine[J]. Talanta,2010,81(3):786-791.
[16]Phansi P,Henrr′quez C,Palacio E,et al Automated in-chip kinetic-catalytic method for molybdenum determination[J]. Talanta,2014,119(15):68-74.
[17]鲍所言,李书存,王桂花,等.催化荧光法测定痕量钼[J].河北大学学报:自然科学版,2001,21(1):61-64.
[18]宋桂兰,郭英,任白皋.液膜分离富集二甲氧基羟基苯基荧光酮荧光猝灭法测定微量钼的研究[J].分析试验室,2005,24(5):44-47.
[19]李英杰,陈世界,高立娣.SAF-Tween80-β-CD 荧光熄灭法测定微量钼的研究[J].分析试验室,2008,27(1):31-33.
[20]王丽红,张辉,李德玲,等.催化荧光光度法测定痕量钼(Ⅳ)[J].理化检验-化学分册,2009,45(9):1072-1074.
[21]李志孙,启文,刘继森,等.甲醇制烯烃催化剂失活模型[J].应用化学,2014,31(4):394-399.
