紫苏油中的多环芳烃残留量的测定方法研究
2015-03-06胡浩斌张小伟张鹏会
胡浩斌,张小伟,张鹏会
(陇东学院化学化工学院,甘肃庆阳 745000)
以不同方法提取的食用紫苏油为研究对象,采用超声波辅助溶剂提取、固相萃取、净化等方法提取油中的多环芳烃,采用气相色谱-质谱-选择离子检测法(GCMS-SIM),对紫苏油中多环芳烃进行检测和分析,为科学合理地优化油脂提取和精炼工艺、检测与控制PAHs、确保食用紫苏油的质量安全具有指导意义。
1 材料与方法
1.1 材料与仪器
采自甘肃省正宁县永正乡的紫苏籽,经干燥、除杂、破碎后,分别采用机械压榨法、正己烷浸提法和超临界CO2流体萃取法提取紫苏油,供分析用。
萘、苊烯、苊、芴、菲、蒽、荧蒽、芘、苯并[a]蒽、屈、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、茚并[1,2,3-c,d]芘、二苯并[a,h]蒽和苯并[g,h,i]芘等16 种组分的混合标准溶液(质量浓度均为10 mg/L,纯度≥99%,北京百灵威科技有限公司);正己烷和二氯甲烷(色谱纯,天津四友精细化学品有限公司);无水硫酸钠(化学纯,用前在420 ℃下干燥4h);其他试剂等均为分析纯(西安化学试剂厂),实验用水符合GB/T6682 一级水要求;硅胶(200 目,青岛胜海化工有限公司,用前在200 ℃下干燥2h);中性氧化铝(60 目,上海纳辉干燥试剂厂,用前在200 ℃下干燥5 h)。
GC6890N/MSD5973N 型气相色谱-质谱联用仪(美国安捷伦科技有限公司),配7683 型自动进样器;KQ-250 型超声清洗仪(昆山市超声仪器有限公司);XK96-B 型快速混匀器(姜堰市新康医疗器械有限公司)。自制硅胶-氧化铝复合层析柱(在30 cm × 1.5 cm 的玻璃柱中,由下往上依次填入5 g 硫酸钠、5 g 硅胶、1 g 氧化铝和5 g 硫酸钠。
1.2 检测条件
色谱条件:HP-5MS 色谱柱(30 m × 0.25 mm ×0.25 μm);进样口温度300 ℃;程序升温:70 ℃保持2 min,25 ℃/min 升至150 ℃,3 ℃/min 升至200 ℃,再以8 ℃/min 升至280 ℃保持10 min;载气为高纯He(99.999%)。恒流模式,流速1.0 mL/min,进样量1.0 μL,脉冲不分流模式进样1 min 后开阀。
质谱条件:电子轰击离子源(EI)温度230 ℃,电子能量70 eV,双四极杆温度150 ℃;传输线温度280℃;猝灭气(He)流量为2.25 mL/min,碰撞气(N2)流量为1.5 mL/min,扫描范围(m/z)50~450 amu,溶剂延迟4 min。
1.3 试验方法
1.3.1 PAHs 的分离和测定 按项1.2 下条件,对16种PAHs 的标准溶液进行扫描,得总离子流图(图1)。根据各化合物的保留时间和质谱图,确定选择离子监测的采集时间和特征离子,并以信噪比为3 (S/N=3)时的对应浓度作为方法的检测限,结果见表1。

图1 标准溶液中16 种PAHs 的总离子流

表1 16 种PAHs 的保留时间、特征离子和检测限
1.3.2 标准工作曲线的绘制 准确移取10 mg/L 的混合标准液0.01、0.02、0.05、0.1、0.5、1.0 mL,分别置于10 mL 容量瓶中,用正己烷定容,得到浓度分别为10、20、50、100、500、1 000 μg/L 系列标准工作溶液。再按照“1.2”下的条件,对系列标准工作液进行GC分析,每个质量浓度水平测定3 次,取平均值。以各组分的峰面积(Y)对质量浓度(X)绘制标准曲线,得到各待测组分标准曲线的线性回归方程和相关系数,结果见表2。

表2 各PAHs 的线性范围、线性方程和相关系数
1.3.3 样品的处理与测定 分别将压榨法、正己烷浸提法和超临界CO2流体萃取得到的紫苏油准确称取5.0 g,置于25 mL 试管中,加入10 mL 正己烷-丙酮(1∶1,V/V),振荡混匀,以250 W 功率超声10 min,得提取液。将硅胶-中性氧化铝层析柱先用15 mL 正己烷-二氯甲烷(1∶1,V/V)混合液活化后,分别加入上述提取液,先用10 mL 正己烷冲洗并弃去冲洗液,再用20 mL正己烷-二氯甲烷(1∶1,V/V)混合液洗脱,洗脱液于40 ℃下自然挥发浓缩至1.0 mL,用正己烷定容至1.0 mL。再按照“1.2”下的检测条件,分别进行测定,总离子流图见图2。
定性检测的依据:被测样品色谱峰的保留时间与标准工作溶液的保留时间相一致;被测样品检测离子的相对丰度与标准工作溶液的相对丰度两者之差不大于允许相对偏差,则可判断样品中存在对应的被测物[1]。
1.3.4 计算方法 根据样品峰面积,由各回归方程计算各PAH 的质量m,再按关系式C=m/W 计算样品中各PAH 的含量C。其中:C 的单位为μg/kg、m 的单位为ng、W 为样品质量,单位为g。
2 结果与讨论
2.1 原料的选取及加工
造成食用油中PAHs 含量高的主要因素为原料及加工污染。为了避免加工过程中引入PAHs,选料时,应考虑:①采用人工收获油料,尽量减少在机械收获、运输、加工等过程中与污染源接触;②用玻璃瓶盛装油料;③选择合适的油料干燥方式并控制合理的水分含量;④筛选环保浸提溶剂,控制溶剂中PAHs的含量。

图2 紫苏油中16 种PAHs 的总离子流图
2.2 色谱条件的选择
色谱分离时,不仅载气流速影响分离度,而且升温程序的不同也会导致色谱分离度的变化。本实验采用混合标准溶液进行色谱条件的优化,最后确定的色谱条件见“1.2”。
2.3 提取方法及条件的选择
由于PAHs 的结构比较稳定,采用超声波提取方法简单、速度较快、提取率高,目前是食品中提取PAHs 大多采用的方法[2]。本试验选用5 种溶剂系统,分别对超临界CO2萃取油品进行超声提取,并按“1.3.3”下的程序进行净化与测定,结果见表3。
从表3 可看出,用正己烷-丙酮(1∶1,V/V)作提取溶剂,除芴、菲、二苯并[a,h]蒽、苯并[g,h,i]芘外,其他PAHs 的提取效率最高。同样,以PAHs的提取效率为指标,考察溶剂用量、超声功率和超声时间对PAHs 提取效率的影响。通过优化得到最佳提取条件为:提取溶剂为正己烷-丙酮(1∶1,V/V)10 mL、超声功率为250 W、超声时间为10 min。

表3 提取溶剂对PAHs 提取效果的影响 单位:μg/kg
2.4 纯化方法的选择
由于PAHs 是非极性或弱极性物质,植物油品基体复杂,PAHs 含量低,稳定性差,而且油脂中含有的大量甘油三酯和脂肪伴随物等干扰因素多,分析难度大。因此,需要对油品进行预处理以富集待测组分,并采用适当方法净化以消除基体的干扰、提高检测的灵敏度,降低检测限。
本试验按照“1.3.3”下的净化方法,以正己烷-二氯甲烷为洗脱剂,分别选用3 种不同的吸附剂对超临界CO2萃取油品进行净化后,再按“1.2”下的条件进行测定,计算各PAHs 的含量,比较各固相萃取柱的净化效果,结果见表4。

表4 吸附剂对PAHs 提取效果的影响 单位:μg/kg
(续)
结果表明,硅胶—中性氧化铝复合柱(5g/1g)的净化效果最好,基质干扰也较少。硅胶价格便宜、净化速率快,但对油品中色素的去除效果不佳;氧化铝对脂肪具有更大的柱容量,且对某些成分有一定的分解作用,洗脱时间长,重现性较差,故不能单独使用氧化铝作填料。经优化选择硅胶-中性氧化铝(5g/1g)为吸附剂,正己烷-二氯甲烷(1∶1,V/V)为洗脱剂对油品进行净化。
2.5 方法学考察
2.5.1 线性关系及检出限 表2 的试验数据表明,在10~1 000 μg/L 浓度范围内,该方法的线性关系良好,相关系数为0.994~0.999,以3 倍信噪比计算的各PAHs 检测限见表1。
2.5.2 精密度和准确度 取1 μL 质量浓度为500 μg/L
的PAHs 混合标准液,在1 d 内按照实验条件连续测定10 次,通过峰面积计算的日内RSD 在3.20%~6.92%范围内。为考察方法的准确度,准确称取5.0 g 已知各组分含量的超临界CO2萃取的紫苏油样品,分别加入质量浓度为500 μg/L 的PAHs 混合标准工作液100 μL、300 μL 和500 μL,每个添加水平做5 个平行样品,按照“1.3.2”下的方法进行提取、净化后,再按“1.2”下的检测条件进行分析,计算各PAH 的回收率和RSD,结果见表5。
由表5 可见,16 种PAHs 的回收率在77.4%~91.7%,RSD 在3.12~8.73% (n=5)。
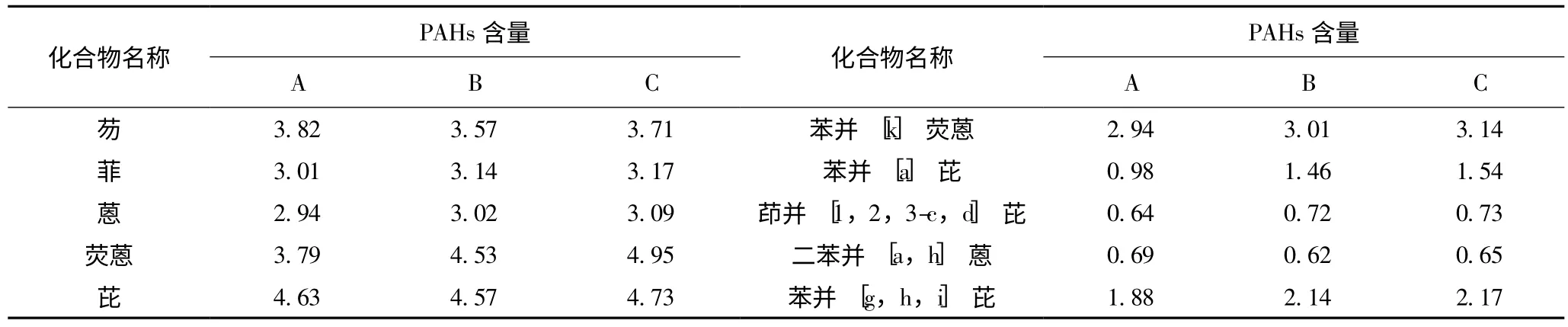
2.6 样品测定结果
用本文所建立的方法测定了3 种紫苏油品中各PAHs 的含量,结果见表6。结果表明,3 种紫苏油中,以萘、苊、二氢苊、荧蒽、芘的含量最高,这与焦化废水和环境空气样品研究的结果有相似性,可见在紫苏生长期,水和大气等环境污染是造成紫苏中PAHs 残留的主要因素;同时,紫苏油中PAHs 的含量随提取方法的不同而有差异,其中超临界CO2流体萃取油品中PAHs的含量最高,而压榨油品中PAHs 的含量最低,但均未超出国家规定的食用油中多环芳烃含量标准[1]。这可能是因为PAHs 是非极性物质,具有脂溶性,采用正己烷浸提或超临界CO2流体萃取法,能使更多的PAHs 被提取出来,而压榨油则更多地留在籽粕中。

表5 16 种PAHs 的加标回收率和精密度(n=5)
3 结论
本文采用GC-MS-SIM 法对用不同方法提取的3 种紫苏油中的16 种PAHs 进行定性和定量分析。在10~1000 μg/L 浓度范围内,16 种PAHs 的线性关系良好,相关系数为0.994~0.999,加标回收率在77.4%~91.7%之间,日内(n=10)RSD 为3.20%~6.92%,检测限为0.08~0.45 μg/kg。溶剂浸提法和超临界CO2萃取法虽然比压榨法的出油率高,油料资源的利用率高,但PAHs 的残留量明显的高于压榨法。该方法快速准确、PAHs 的净化和分离度好、分析灵敏度高、重复性好,可适用于对紫苏油中PAHs 的快速分离与检测。食用油中PAHs 的污染已经引起世界各国的广泛重视[3-5]。为确保食用油脂质量安全,针对PAHs 的来源,食用油生产企业要从原料的收获、干燥、运输等过程抓起,选用科学合理的油脂提取和精炼工艺,开发简单、快速的食用油中PAHs 检测方法,加强生产和储藏过程中PAHs 的实时、在线监控,避免有害因素的形成,确保食用油安全。

表6 紫苏油中各PAHs 的含量
[1]中华人民共和国国家标准GB/T 23213-2008.植物油中多环芳烃的测定——气相色谱-质谱法,3-4.
[2]章汝平,何立芳.食品中多环芳烃的提取、纯化以及检测方法的研究进展[J].食品科技,2007,24 (1):20-25.
[3]张根生,赵全,岳晓谧,等.食品中有害化学物质的危害与检测[J].中国计量,2006,24 (6):222-230.
[4]张小涛,刘玉兰,王月华.食用油脂中多环芳烃的研究进展[J].中国油脂,2012,37 (10):45-49.
[5]Moret S,Purcaro G,Conte L S.Polycyclic aromatic hydrocarbons in vegetable oils from canned foods[J].Eur.J.Lipid Sci.Technol.,2005,107 (7-8):488-496.
