CoSb3基方钴矿化合物的Ga,In掺杂及相关复杂缺陷的研究
2015-02-25席丽丽邱雨婷陈立东杨继辉张文清
席丽丽,邱雨婷,史 讯,杨 炯, 陈立东,杨继辉,张文清
(1.中国科学院上海硅酸盐研究所,上海 200050) ( 2.美国华盛顿大学,西雅图 98195-2120)

CoSb3基方钴矿化合物的Ga,In掺杂及相关复杂缺陷的研究
席丽丽1,邱雨婷1,史讯1,杨炯2, 陈立东1,杨继辉2,张文清1
(1.中国科学院上海硅酸盐研究所,上海 200050) ( 2.美国华盛顿大学,西雅图 98195-2120)
摘要:通过第一性原理与热力学结合的方法,研究了Ga, In等掺杂的CoSb3基方钴矿化合物中的复杂缺陷问题。详细计算了Ga,In在CoSb3中填充,Co,Sb位置替换以及填充-替换同时共存等缺陷的形成能。研究结果表明, 缺陷形成能与费米能级、化学势等相关。Ga,In等在方钴矿中不是单纯的填充,而是填充和Sb位置替换同时共存的复杂缺陷。Ga掺杂以填充-替换比例2∶1的缺陷为主,而In掺杂,根据不同的条件可形成填充, 替换, 以及不同比例的填充替换复合缺陷, 其中尤其以4∶2和2∶1最多。根据巨正则系宗, 研究了Ga, In掺杂系统的载流子浓度和各缺陷的浓度。 发现Ga, In掺杂的方钴矿由于填充和替换电荷的自补偿效应, 其载流子浓度较低,尤其是Ga填充, 具有类似本征半导体的低载流子浓度,且得到实验证实。In掺杂系统由于填充替换的比例偏离2∶1, 填充位置的In比Ga的稍高一些,因此具有比Ga掺杂更高的载流子浓度。
关键词:方钴矿;第一性原理;热电材料;复杂缺陷结构
1前言
在能源和环境问题日益加剧的今天,热电材料作为一种绿色环保的新型能源材料而受到广泛关注[1-4]。热电转换技术利用半导体材料的赛贝克效应和帕尔贴效应实现热能和电能之间的直接转换[1-2]。这种技术可将许多废弃的热能,例如汽车尾气废热和工业余热等,直接转换为电能而提高能源利用率。从19世纪热电效应发现到上世纪90年代中期,热电研究主要集中于传统的Si-Ge合金[5]和以Bi2Te3与PbTe为基础的窄带半导体中[6-8],热电优值ZT在1.0左右徘徊。目前应用广泛,技术发展比较成熟的Bi2Te3固溶体合金材料,其稳定的热电优值ZT值只有1.0左右,器件的转换效率低于5%,远低于传统热机转换效率,只在一些特殊的制冷及太空电源中应用。有效提高热电材料转换效率成为近几年来热电材料研究的热点与难点。
热电材料的能量转换效率由材料的热电优值ZT值决定,ZT值越高转换效率越高。ZT(=S2σT/κ)值决定于相互关联的三个物理参数,其中S为塞贝克系数,σ为电导率,κ为热导率(包含电子热导率κe和晶格热导率κL两部分)。这3个物理参数之间互相关联,通常塞贝克系数的提高伴随着电导率的减少,反之亦然。20世纪90年代美国科学家Slack G提出“声子玻璃-电子晶体”(Phonon Glasses Electron Crystal, PGEC)的设计概念[9-11],发现了一些新型笼状结构,以方钴矿和Clathrates为代表,开创了热电材料研究的新方向[12-14]。
方钴矿化合物是一类有发展前景的新型笼状热电化合物[15]。方钴矿最大的特点是其晶体结构中有两个由12个Sb原子形成的二十面体孔洞,该孔洞可以填入一些杂质原子,这些杂质原子可以是碱金属,碱土金属,以及稀土金属。具体能否填入孔洞与填充原子的电负性和Sb原子电负性之差有关系,即电负性选择规则(xI-xSb>0.8)。填充原子可以优化载流子浓度,同时在孔洞中振动散射声子,降低晶格热导率,从而优化热电性能,使得方钴矿材料的热电性能从单填的1.0,到双填的1.2,再到多填的1.7,使其成为目前最具发展潜力的热电材料之一[16-28]。这些元素在方钴矿中的掺杂形态比较简单,基本上都是占据晶格孔洞位置,其填充对方钴矿材料热电性能的进一步提升效果不明显。对于一些不满足电负性选择规则的元素,比如ⅢA元素Ga,In,由于他们的半径较小,在方钴矿中的存在形式一直存在争议。理论上,系统的研究这些元素在方钴矿中的掺杂,对方钴矿化合物结构设计和性能优化具有重要的意义。
虽然根据Shi等人的结论,Ga,In等不能在方钴矿晶格孔洞中稳定存在,在实验上对Ga,In等在方钴矿化合物中掺杂及其对性能做了一些研究,且得到较好热电性能的Ga,In掺杂方钴矿热电材料。Harnwunggmoung 等人研究了Ga掺杂的CoSb3基热电材料[29],发现只有少量的Ga能填入方钴矿的晶格孔洞中。He和 Mallik 等人报道了In掺杂的CoSb3基方钴矿[30-32],发现In在CoSb3晶格孔洞中的填充量上限为0.22。Grytsiv 等人发现In在CoSb3中填充量上限与系统初始元素化学配比有关,在与CoSb2和InSb共存时,其最大填充量为0.22,而富Sb情况下,其最大固溶度为0.09[33]。Sesselmann等人发现在In掺杂的方钴矿中,In也有可能占据Sb位置,形成取代Sb的杂质缺陷[34-35]。除了这些元素的单掺杂外,研究人员对Ga,In等与其他元素共填的方钴矿结构进行研究,并获得了较好的热电性能。这些研究虽然没有确定Ga,In在方钴矿中真实的存在形式,但发现了Ga,In在方钴矿中和其他填充原子不同,并不是单纯的占据孔洞位置。由于Ga3+与In3+离子半径与Co3+类似,因此Ga,In也有可能取代Co位置。
最近,作者通过理论与实验的系统研究[36-38],发现Ga,In在方钴矿中存在形式比较复杂,倾向于形成晶格孔洞位和Sb位置共存的复杂缺陷形态。通过第一性原理与热力学结合的方式,我们系统研究了Ga,In在方钴矿中可能存在的各种缺陷及其形成能,通过巨正则系综理论研究了不同缺陷的缺陷浓度和系统的载流子浓度,并对形成复合缺陷的原因进行了分析。
2计算方法
2.1能量计算
第一性原理计算采用了基于密度泛函理论的Vienna Ab initio Simulation Package (VASP)程序包[39-40]。所有计算均采用缀加平面波(PAW)的方法来处理电子波函数,而交换关联势选用广域梯度近似(GGA)下的Perdew-Burke-Ernzerhof (PBE) 形式。平面波截断能取320 eV,电子自洽能量收敛标准为10-4eV,所有原子受力均小于0.01 eV。因为方钴矿化合物每个晶胞有两个孔洞,为了系统的研究,计算中我们采用2×2×2的超胞,这样未填充的方钴矿有128个原子和8个孔洞。VASP采用Monhkorst-Pack特殊网格点方法,对于方钴矿化合物采用了3×3×3个K点,其他的化合物采用15×15×15的K点。计算中对理想的超胞对晶格常数和离子位置都做了弛豫。对于含缺陷的体系,保持晶胞体积不变,对离子位置进行弛豫。对于缺陷的不同构型尤其是复合缺陷构型较多,通过计算选取能量最低的构型作为进一步计算的依据。
2.2形成能的计算
本文中,我们考虑了Ga,In掺杂的CoSb3化合物的各种缺陷,包括填充位置缺陷MVF, 替换缺陷MSb,MCo,和其他的复合缺陷 MVF(0, +1), MSb(0,-1,-2, +1), MVF-MSb(0, -1, +1), 2MVF-MSb(0,-1, +1), 3MVF-MSb(0, +1), 4MVF-MSb(0, +1, +2), 4MVF-2MSb(0), 5MVF-2MSb(0, +1), 和 6MVF-3MSb(0)。括号中数字为缺陷的价态。一些本征缺陷如Co,Sb空位,间隙等也考虑其中,由于MCo缺陷形成能较高,因此之后的研究没有仔细考虑。由于本征缺陷Coi形成能与其他杂质缺陷的形成能可比,因此该缺陷也详细的研究[41]。
本文中复杂缺陷MxCo32-zMzSb96-yMy的以(D,q)来标记,其缺陷形成能ΔGf(D,q)可以写为以下形式[42-44]:
ΔGf(D,q)=[Etot(D,q)-Etot(bulk)]+yμSb+z μCo-(x+y+z)μM+q(εF+Ev+ΔV)]/(x+y+z)
(1)
2.3缺陷浓度的计算
根据巨正则系宗热力学模型研究不同缺陷的浓度问题[43-44]。给定一个超胞,其中填充位置数为N,则Co和Sb位置的占据数分别为4N和12N。ni代表缺陷i的数目,m是总的缺陷类型。本征缺陷Co间隙位的缺陷表示为Coi数目记为n0。 而其他的杂质缺陷MVF, MSb, MVF-MSb, 2MVF-MSb, …,aMVF-bMSb分别标记为 n1(1-0), n2(0-1), n3(1-1), n4(2-1),… , 和 nm(a-b)。 a,b分别为该缺陷中占据孔洞和Sb位置的数目。当我们取得超胞足够大,不同缺陷之间的相互作用可以忽略,则整个系统的吉布斯自由能可写为:
J=U-TS-NMμM-NSbμSb-NCoμCo
(2)
其中U为系统的内能,NM,NSb, 和NCo分别为系统中孔洞,Sb位,Co位的晶格数。假设系统同时存在上述的一些缺陷,则
(3)
NCo=4N+n0
(4)
(5)
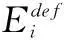
(6)
(7)
其中
(8)
通过对自由能分别对缺陷i求导,则可得到不同缺陷的缺陷浓度。约化后不同缺陷的缺陷浓度写为以下形式:
C0=n0/N≈3exp(-g0/kT)
C1(1-0)=n1/N≈exp(-g1/kT)
C2(0-1)=n2/N≈12exp(-g2/kT)
C3(1-1)=n3/N≈12exp(-g3/kT)
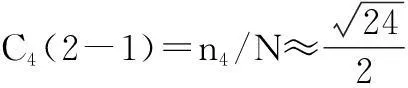
......

exp(-gi/(aikT))
(ai×12-24)1/aiexp(-gi/(aikT))
(9)
其中费米能级可以通过下面的电中性条件得到。

(10)
其中q(i) 是缺陷i所带的电荷。Ns是缺陷i的单位体积的个数。ne和nh分别是系统中电子和空穴浓度。
3结果和讨论
3.1缺陷形成能
通过前述缺陷形成能的计算模型,我们计算了Ga,In掺杂的方钴矿体系中各种不同缺陷的形成能。表1给出了Ga,In掺杂的CoSb3体系中,不同缺陷在富Sb和富Co情况下的缺陷形成能,其中CoSb3化合物的本征缺陷形成能也列在表中做对比。从表1中可以看出,相较于杂质缺陷,CoSb3中的本征缺陷的形成能都较高,只有Co的间隙位置Coi的形成能稍低一些。因此我们考虑了这一缺陷。其它的杂质缺陷除去MVF,MSb等单填充和单Sb位置替换外,填充和Sb替换组合成的复杂缺陷形成能较低,因此主要详细研究了这些缺陷的形成能以及其对应的缺陷浓度。

表1 0 K下,Ca,In 掺杂的CoSb3 体系中不同缺陷在富Co和富Sb情况下的缺陷的形成能(eV)
Note:Superscript a representing data from Parketal.,PhysRevB,2010,81:085 206
图1是1 000 K 下Ga,In掺杂方钴矿体系在富Sb和富Co情况下,一些主要的缺陷形成能和费米能级的关系。从图中可以看出,随着费米能级的变化,不同缺陷的形成能相对大小发生变化。对Ga掺杂的系统,不管是富Sb还是富Co的情况下,均主要形成填充和Sb替换同时存在,比例为2∶1的缺陷。只有在费米能级很高的情况下,形成Ga替换Sb的取代缺陷。对于In掺杂的方钴矿化合物,情况要复杂一些。在不同的费米能级下,可以形成纯填充,填充与替换共存,以及纯Sb取代的不同缺陷。同时可以看出,不同比例的双位缺陷形成能非常接近,表明这些复杂缺陷可以同时共存于In掺杂的方钴矿体系中。这些结果与之前实验结果一致,Ga在方钴矿中以2∶1的比例占据孔洞位置和Sb位置,Ga在孔洞位置显示+1价,而在Sb取代位置刚好比Sb少两个电子,形成一种电荷自补偿的复合缺陷,由于这种电荷自补偿效应,其化合物具有很低的载流子浓度,形成类似于本征的半导体的化合物。前面也提到In掺杂在CoSb3中,情况比较复杂,不同的实验条件,得到的结果也不尽相同,这和我们的计算结果比较符合[37]。
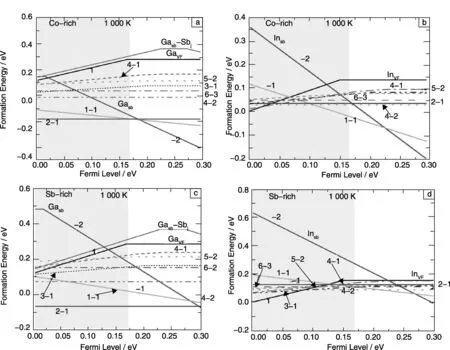
图1 1 000 K下,单位杂质原子的Ga,In掺杂的方钴矿CoSb3缺陷形成能与费米能级的关系:(a,b)为富Co的情况,(c,d)为富Sb的情况Fig.1 Defect formation energies of CoSb3 doped by Ga,In (per impurity atom) as a function of Fermi level:(a,b) Co-rich situation;(c,d) Sb-rich situation
3.2缺陷和载流子浓度
通过缺陷形成能的研究发现,Ga,In掺杂的方钴矿体系中,缺陷比较复杂,不同类型的缺陷可同时存在。用巨正则系宗的热力学模型,在电中性条件下,估算Ga,In掺杂下方钴矿体系存在的不同缺陷浓度。图2显示了单位体积内不同缺陷的缺陷浓度和载流子浓度与Sb有效化学势的变化关系。图2以红色的竖线分成两部分,其中左边是杂质原子Ga,In有效化学势为0的情况,右边是避免生成杂质相MSb的M有效化学势取值。虽然理论上Sb的有效化学势范围可以在-0.33~0 eV之间变化,但是实验上元素化学势控制是非常困难的,实验上一般都是富含Sb或者缺少量Sb的情况,因此实验结果主要落在图中右边的一小部分。从图2中可以看出Ga掺杂的方钴矿体系,在整个范围内主要的缺陷是电荷自补偿的双位掺杂复合缺陷,而其他缺陷浓度相比该缺陷都较低。在电中性条件下,Ga掺杂方钴矿只在Sb化学势很低(<-0.28 eV)情况下为弱p型,其他范围内均为弱n型,且最大载流子浓度为2×1019cm-3,这和之前的结果比较吻合[36]。
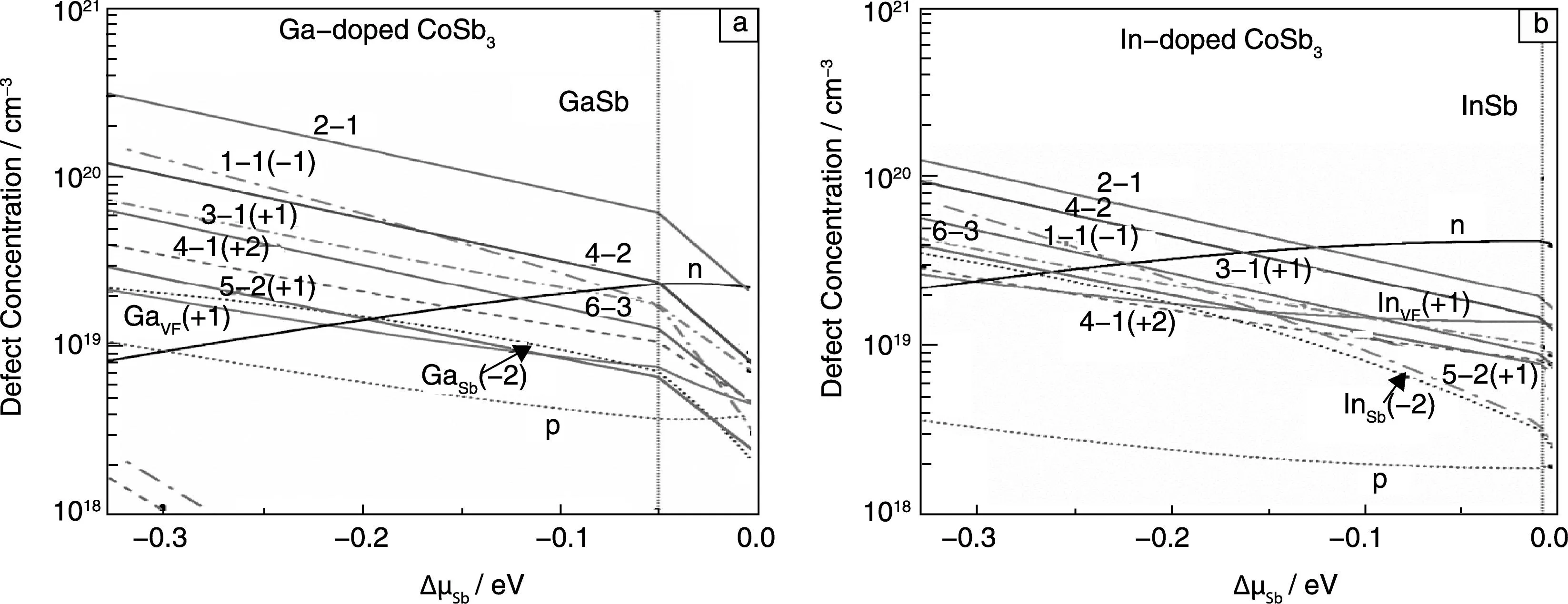
图2 单位体积不同缺陷的缺陷浓度和Sb有效化学势的关系及电子和空穴浓度:(a)Ga掺杂,(b) In 掺杂Fig. 2 Equilibrium defect concentrations for Ga- and In- doped CoSb3 as a function of Sb chemical potential and the calculated concentrations of electrons and holes per unit volume at 1 000 K :(a) Ga-doped CoSb3,(b) In-doped CoSb3
对于In掺杂的系统,可以看出除了双位复合缺陷(填充和替换位置比例为2∶1),双位掺杂缺陷浓度(比例为4∶2)也较高,而其他的双位掺杂缺陷浓度都很接近,这和之前形成能的计算结果也符合。除了这些复杂的双位复合缺陷,In在孔洞位置的单填充缺陷浓度也比Ga的高。由于In在孔洞中提供的电子为n型掺杂,因此In掺杂的系统在整个Sb化学势变化范围内均为n型掺杂,载流子浓度也比Ga掺杂系统的高,最高达到4.3×1019cm-3,这个值比最近的实验值2×1020cm-3要稍低一些,这可能是因为实验制备的复杂性和多样性,存在一些不可预测的缺陷。
虽然理论上可以算出各个不同缺陷的缺陷浓度,但是实验上测试条件的限制,一般只能测出制备出的材料的整体结构特征,比如Ga,In等在孔洞位置和在Sb位置总浓度,而想要细分每种缺陷各占的百分比却比较困难,因此计算了Ga,In在方钴矿中总的固溶度以及其在孔洞中和在Sb位置总的固溶度,如图3所示。图3纵坐标为掺杂原子与总空洞数的相对浓度,从图中可以看出,在Sb化学势为0时,Ga在空洞中和在Sb位置的总浓度为0.12,与之前的实验值比较吻合,随着Sb化学势的降低,Ga在方钴矿中的固溶度逐渐增加。而通过比较Ga在空洞中的固溶度和在Sb位置固溶度时发现,在整个范围内,Ga在两个位置总体比例基本上为2∶1,这也从侧面说明Ga在方钴矿中以2∶1比例存在于孔洞位置和Sb取代位置,形成完全的电荷自补偿的复合缺陷。在Sb化学势较低的情况,Ga在方钴矿中的固溶度非常高,在空洞中达到0.9,但是由于实验上Sb化学势控制的难度,这一高掺杂量的化合物在实验上很难实现。

图3 Ga,In在方钴矿中的总的固溶度和它们在填充位置及Sb取代位置的固溶度与Sb有效化学势的关系:(a) Ga,(b) InFig.3 Total impurity atom concentration and their filling position and Sb replaced position as a function of Sb chemical potential:(a) Ga-containing CoSb3 and (b) In-containing CoSb3
对于In掺杂的系统,In在填充孔洞位置和在Sb位置的比例高于2∶1,说明在In掺杂系统中,不是完全的电荷补偿,在孔洞中的要稍高一些。这也和之前的理论和实验结果一致。在Sb化学势为0时,In在方钴矿中的总固溶度0.2,这比最近实验上固溶度最大0.27稍低一些,但是之前的讨论表明,固溶度与Sb化学势密切相关,虽然实验上控制困难,但是一般是与CoSb3中化学配比相关,由于In在Sb位置稍多一些,所以In掺杂系统的Sb化学势应该比Ga掺杂的系统低,通过控制Sb化学势,发现,在Sb化学势为-0.05 eV时,In在方钴矿中的固溶度为0.272,与实验值接近。在Sb化学势更低时,In的固溶度也逐渐增加,理论上最大可达到在空洞中的固溶度为0.7。
3.3材料微结构特性和化学键
通过讨论,得出Ga,In等掺杂元素与其他的满足电负性选择规则的元素不同,这些元素都形成稳定单填充的方钴矿化合物,而Ga,In很难形成单一的填充或者替换的缺陷,而是倾向于形成填充-替换同时存在的复合缺陷。为了验证上述结果的正确性,通过SEM和TEM分析了成分为(GaVF)0.1Co4Sb11.95(GaSb)0.05化合物不同位置的微结构,得出整体的成分组成[38],如图4所示。图4a中三角图形为SEM结果,圆圈图形为TEM结果,图4b中三角图形为TEM结果,圆圈图形为SEM结果,图中斜线为理想的掺杂原子在填充和替换位置比例为2∶1时Sb原子百分数随Ga,In原子百分数的变化,从图中可以看出,不管是实验上原始配比如何,还是同一种样品不同位置的微结构成分分析,随着Ga,In量的增多,Sb量线性减少,并沿着理想的斜线mSb=75%-5/6nM(M=Ga, In)。
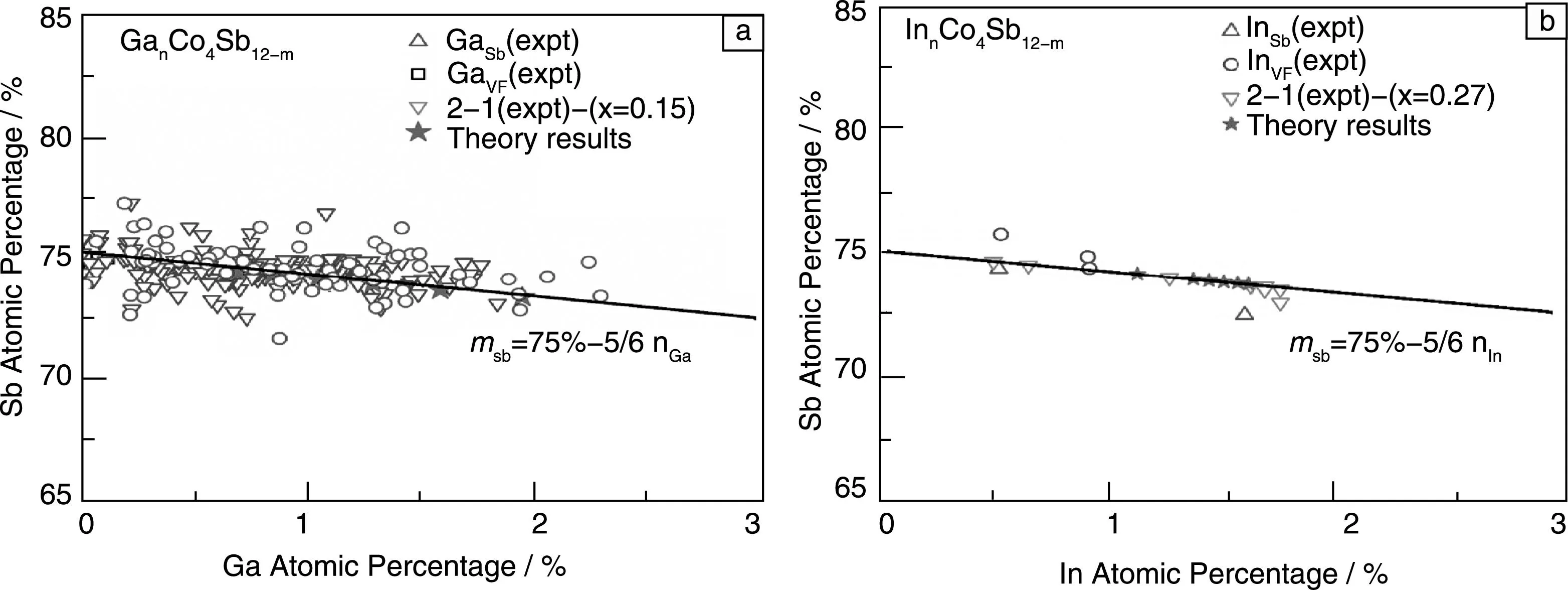
图4 不同成分的Ga,In掺杂的方钴矿化合物中Sb原子百分数随着掺杂原子Ga,In原子百分数的变化:(a) Ga掺杂,(b)In掺杂Fig.4 Sb atomic percentage as a function of Ga,In atomic percentage in Ga-and In-doped CoSb3:(a) Ga doping,(b) In doping
图4从实验上进一步证明了Ga,In等元素在方钴矿中倾向于形成电荷自补偿双位掺杂的复合缺陷,这可能与Ga,In的半径有关,因为Ga,In电子结构类似于碱金属,最外层有一个电子。之前一系列填充量研究的工作表明填充原子半径太大或者太小都不能形成稳定的填充方钴矿化合物。而填充和替换同时存在,一方面电荷补偿的效应使得体系能量降低,另一方面填充原子和周围取代Sb位置的杂质原子之间相互作用也使得系统能量降低。图5给出了正常Ba填充和Ga掺杂的方钴矿中一个Sb四元环和中间孔洞位置的电荷密度差,虚线表示失电子,实线表示得电子。从图中可以看出,对于Ba填充的方钴矿,Ba与周围Sb原子主要形成离子键,填充原子向方钴矿提供电子,而Ga由于离子半径小,偏离孔洞的中间位置,并且与周围的Sb原子和取代Sb位置的Ga原子形成共价键,这一化学键的区别也造成了Ga,In等在方钴矿中形成不同于一般正常填充原子的单一填充缺陷。
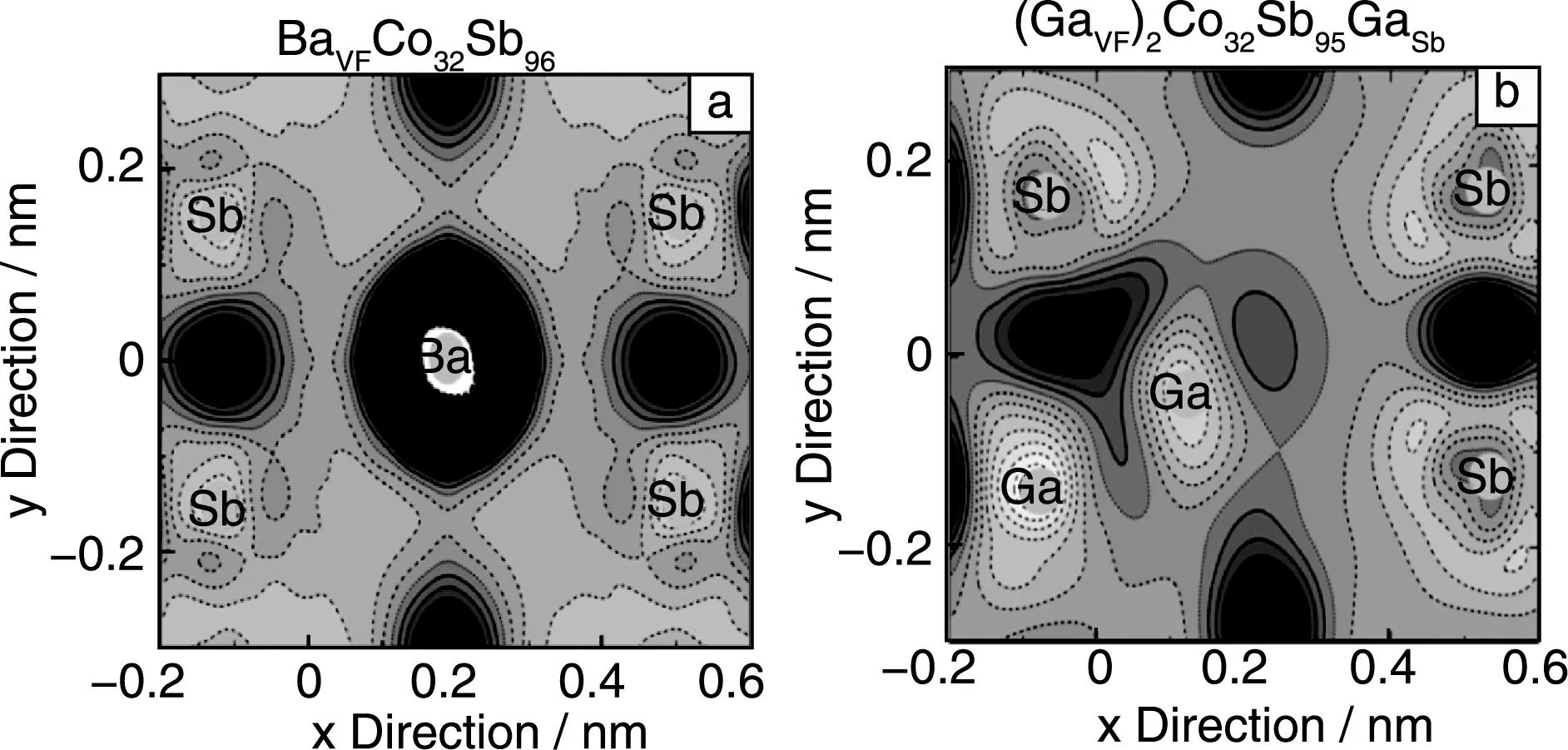
图5 Sb原子四元环的电荷密度差:(a) BaCo32Sb96, (b) Ga2Co32Sb95GaFig.5 Charge density difference at Sb four-ring plane in BaCo32Sb96(a) and Ga2Co32Sb95Ga (b)
3.4Ga,In在方钴矿中的高固溶度及其亚稳特性
图3指出在Sb化学势低的情况下,Ga,In在方钴矿中的固溶度很高,但是这是在高温下的结果,从表1可以看出,所有的缺陷形成能在0 K时都是正值,说明这些缺陷实际上是亚稳的,通过考虑温度带来的构型熵的因素,某些复合缺陷由正值转变为负值。考虑了电荷完全补偿电中性的几个复合缺陷在不同Sb化学势情况下的缺陷形成能,如图6,横坐标表示不同电中性缺陷(填充位置和替换位置的比例分别为2∶1, 4∶2, 6∶3),分别取Sb化学势两个极限0 eV和-0.33 eV。从图中可以看出在低Sb化学势的情况,Ga,In杂质缺陷的形成能在高温下都很低,对于Ga掺杂系统,缺陷2-1为负值,且随着掺杂量的增多,形成能增加并不多,在0 eV左右徘徊,甚至6-3的高掺杂缺陷,能量也很低,对于In掺杂的系统,随着掺杂量的增加,其缺陷形成能几乎没有变化,均表明在高温和Sb低化学势条件下,Ga,In均可形成高固溶度的亚稳掺杂方钴矿化合物。

图6 1 000 K下几个完全电荷补偿缺陷的形成能Fig.6 Formation energies of a few different fully charge-compensated defects under different Sb chemical potentials at 1 000 K
4结论
通过系统研究Ga,In掺杂CoSb3方钴矿化合物中的缺陷形式。结果表明,在0 K下,Ga,In均很难形成稳定的填充或替换,或填充替换同时存在的各种缺陷。高温1 000 K下,考虑了不同原子的构型熵,Ga,In在方钴矿中的缺陷形式与实验条件密切相关。缺陷形成能的研究发现,Ga主要形成填充-替换比例为2∶1的完全电荷自补偿复合缺陷,而在高费米能级时形成Ga替换Sb的取代缺陷。而In原子形成何种缺陷对实验条件更加敏感,在不同费米能级时,可形成单填充,填充-替换,以及单替换的不同缺陷形态。载流子浓度的研究结果表明,Ga,In均为n型掺杂的半导体,由于Ga掺杂方钴矿的主要缺陷是电荷完全补偿的2-1复合缺陷,其载流子浓度很低,几乎接近本征半导体。而In形成的主要缺陷虽然也是2-1,4-2的电荷补偿缺陷,但是由于In在晶格孔洞中的填充缺陷浓度稍高一些,因此具有比Ga掺杂更高的载流子浓度。进一步对化学键的分析表明,Ga,In等倾向于形成复合缺陷的原因是其离子半径较小,在空洞位置会偏离中心位置而与周围的Sb原子和杂质原子形成共价键,降低体系的能量。本工作提供了一个有效的优化热电性能的方法,可以在更大范围内调整载流子浓度,同时双位的掺杂对降低热导率更加有效。这种巨正则系宗研究缺陷浓度的方法也可用于其他材料的缺陷研究中。
参考文献References
[1]Liu Enke (刘恩科),Zhu Bingsheng (朱秉升),Luo Jinsheng (罗晋生).Semiconductorphysics(半导体物理学)[M]. Beijing: Electronic Industry Press,2003.
[2]Ioffe A F.ThermoelementsandThermoelectricCooling[M]. Infosearch, London, UK, 1956:1-33.
[3]Chen G, Dresselhaus M S, Dresseihaus G,etal. Recent Development in Thermoelectric Materials[J].InterMaterRev, 2003, 48(1): 45-66, and references therein.
[4]Nolas G S, Sharp J, Goldsmid H J.ThermoelectricsBasicPrinciplesandNewMaterialsDevelopments[M]. Springer, 2001.
[5]Ni W, W. Chen W M, Buyanova I A,etal. Some Critical Issues on Growth of High Quality Si and SiGe Films Using a Solid-source Molecular Beam Epitaxy System[J].JCrystalGrowth, 1995, 157: 242-247.
[6]Frost R T, Corelli J C, Balicki M. Reactor Irradiation PbTe, Bi2Te3and ZnSb[J].AdvancedEnergyConversion, 1962, 2: 77-78.
[7]Harman T C. Control of Imperfections in Crystals of Pb1-xSnxTe, Pb1-xSnxSe, PbSn1-xSex[J].JNonmetals, 1973, 1: 183.
[8]Gelbstein Y, Dashevsky Z, Dariel M P. High Performance n-Type PbTe-Based Materials for Thermoelectric Applications[J].PhysicaB-Condensedmatter, 2005, 363(1-4): 196-205.
[9]Singh G J. Theoretical and Computational Approaches for Identifying and Optimizing Novel Thermoelectric Materials[M]//RecentTrendsinThermoelectricMaterialsResearch.Amsterdam:Elsevier, 2001, 70: 125-177.
[10]Sales B, Mandrus D, Williams R K. Filled Skutterudite Antimonides: A New Class of Thermoelectric Materials[J].Science, 1996, 272(5 266): 1 325-1 328.
[11]Nolas G S, Morelli D T, Tritt T M. Skutterudites: A Phonon-Glass Electron-Crystal, Approach to Advanced Thermoelectric Energy Conversion Applications[J].AnnuRevMaterSci, 1999, 29: 89-116.
[12]Madsen G K H, Schwarz K, Blaha P,etal. Electronic Structure and Transport in Type-I and Type-VIII Clathrates Containing Strontium, Barium, and Europium[J].PhysRevB, 2003, 68(12): 125 212.
[13]Blake N P, Latturner S, Bryan J D,etal. Band Structures and Thermoelectric Properties of the Clathrates Ba8Ga16Ge30, Sr8Ga16Ge30, Ba8Ga16Si30, and Ba8In16Sn30[J].JChemPhys, 2001, 115(17): 8 060-8 073.
[14]Slack G A.InThermoelectricHandbook[M]. Powe D M (CRC, Boca Raton,FL ),1995:407-440.
[15]Uher C. InRecentTrendsinThermoelectricMaterialsResearchII,SemiconductorsandSemimetals[M]. San Diego: Academic Press, 2000, 69:139-253.
[16]Chen L D, Kawahara T, Tang X F,etal. Anomalous Barium Filling Fraction and N-type Thermoelectric Performance of BayCo4Sb12[J].JApplPhys, 2001, 90(4): 1 864-1 868.
[17]Shi X, Zhang W, Chen L D,etal. Filling Fraction Limit for Intrinsic Voids in Crystals: Doping in Skutterudites[J].PhysRevLett, 2005, 95(18): 185 503 and references therein.
[18]Zhang W, Shi X, Mei Z G,etal. Prediction of an Ultrahigh Filling Fraction for K in CoSb3[J].ApplPhysLett, 2006, 89 (11): 112 105.
[19]Mei Z G, Zhang W, Chen L D,etal. Filling Fraction Limits for Rare-Earth Atoms in CoSb3: An Ab Initio Approach[J].PhysRevB, 2006, 74(15): 153 202.
[20]Xi L, Yang J, Zhang W,etal. Anomalous Dual-Element Filling in Partially Filled Skutterudites[J].JAmChemSoc, 2009, 131(15): 5 560-5 563.
[21]Xi L, Yang J, Zhang W,etal. Filled Skutterudites: from Single to Multiple Filling[J].JKorCeramSoc, 2010, 47(1): 54-60.
[22]Xi L, Yang J, C. F. Lu C F,etal. Systematic Study of the Multiple-Element Filling in Caged Skuterudite CoSb3[J].ChemMater, 2010, 22(7): 2 384-2 394.
[23]Yang J, Zhang W, Bai S Q,etal. Dual-Frequency Resonant Phonon Scattering in BaxRyCo4Sb12(R=La, Ce, and Sr) [J].ApplPhysLett, 2007, 90(19): 192 111.
[24]Shi X, Kong H, Li C P,etal. Low Thermal Conductivity and High Thermoelectric Figure of Merit in N-type BaxYbyCo4Sb12Double-filled Skutterudites[J].ApplPhysLett, 2008, 92(18): 182 101.
[25]Bai S Q, Pei Y Z, Chen L D,etal. Enhanced Thermoelectric Performance of Dual-element-filled Skutterudites BaxCeyCo4Sb12[J],ActaMater, 2009, 57(11): 3 135-3 139.
[26]Zhao W Y, Dong C L, Wei P,etal. Synthesis and High Temperature Transport Properties of Barium and Indium Double-filled Skutterudites BaxInyCo4Sb12-z[J],JApplPhys, 2007, 102(11): 113 708.
[27]Li H, Tang X F, Zhang Q J,etal. High Performance InxCeyCo4Sb12Thermoelectric Materials with in Situ Forming Nanostructured InSb Phase[J].ApplPhysLett, 2009, 94(10): 102 114.
[28]Shi X, Yang J, Salvador J R,etal. Multiple-Filled Skutterudites: High Thermoelectric Figure of Merit through Separately Optimizing Electrical and Thermal Transport[J].JAmChemSoc, 2011, 133(20):7 837-7 846.
[29]Harnwunggmoung A, Kurosaki K, Plirdpring T,etal. Thermoelectric Properties of Ga-Added CoSb3Based Skutterudites[J].JApplPhys, 2011, 110(1): 013 521.
[30]He T, Chen J Z, Rosenfeld H D,etal. Thermoelectric Properties of Indium-Filled Skutterudites[J].ChemMater, 2006, 18(3): 759-762.
[31]Mallik R C, Jung J Y, Ur S C,etal. Thermoelectric Properties of InzCo4Sb12Skutterudites[J].MetMaterInt, 2008, 14(2): 223.
[32]Mallik R C, Mueller E, Kim I H. Thermoelectric Properties of Indium Filled and Germanium Doped Co4Sb12Skutterudites[J].JApplPhys, 2012, 111(2): 023 708.
[33]Grytsiv A, Rogl P, Michor H,etal. InyCo4Sb12Skutterudite: Phase Equilibria and Crystal Structure[J].JElectronMater, 2013, 42(10): 2 940-2 952.
[34]J. Graff J, S. Zhu S, T. Holgate T,etal. High-Temperature Thermoelectric Properties of Co4Sb1-Based Skutterudites with Multiple Filler Atoms: Ce0.1InxYbyCo4Sb12[J].JElectronMater, 2011, 40(5): 696-701.
[35]Sesselmann A, Dasgupta T, Kelm K,etal. Transport Properties and Microstructure of Indium-Added Cobalt-Antimony-Based Skutterudites[J].JMaterRes, 2011, 26(15): 1 820-1 826.
[36]Qiu Y T, Xi L, Shi X,etal. Charge-Compensated Compound Defects in Ga-Containing Thermoelectric Skutterudites[J].AdvFuncMater, 2013, 23(25): 3 194-3 203.
[37]Tang Y L, Qiu Y T, Xi L,etal. Phase Diagram of In-Co-Sb System and Thermoelectric Properties of In-containing Skutterudites[J].EnergyEnvironSci, 2014, 7(2): 812-819.
[38]Qiu Y T, Xing J J, Gao X,etal. Electrical Properties and Microcosmic Study on Compound Defects in Ga-contained Thermoelectric Skutterudites[J].JMaterChemA, 2014, 2(28): 10 952-10 959.
[39]Kresse G, Furthmuller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set[J].PhysRevB, 1996, 54(16): 11 169-11 186.
[40]Kresse G, Hafner J. Ab Initio Molicular-Dynamics for Liquid-Metals[J].PhysRevB, 1993, 47(1): 558-561
[41]Park C H, Kim Y S. Ab Initio Study of Native Point-Defects in CoSb3: Understanding Off-stoichiometric Doping Properties[J].PhysRevB, 2010, 81(8): 085 206.
[42]Zhao J L, Zhang W Q, Li X M,etal. Convergence of the Formation Energies of Intrinsic Point Defects in Wurtzite ZnO: First-Principles Study by Projector Augmented Wave Method[J].JPhys:Conden.Matter, 2006, 18(5): 1 495-1 508.
[43]Nazarov R, Hickel T, Neugebauer J. First-principles Study of the Thermodynamics of Hydrogen-Vacancy Interaction in Fcc Iron[J].PhysRevB, 2010, 82(22): 224 104.
[44]Zhang W, Smith J R, Wang X G. Thermodynamics from Ab Initio Computations[J].PhysRevB, 2004, 70(2): 024 103.
(编辑:盖少飞易毅刚)
Complex Doping of Ga, In and Related Defect Studyin Caged Skutterudites CoSb3
XI Lili1, QIU Yuting1, SHI Xun1, YANG Jiong2,
CHEN Lidong1, YANG Jihui2, ZHANG Wenqing1
(1.Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China)
(2.University of Washington, Seattle WA 98195-2120, USA)
Abstract:Complex doping is one of the challenging problems to be understood in doping and the associated structure tuning in materials science. Here we investigate the single-impurity-induced complex doping behaviors of group 13 elements Ga or In in caged skutterudite CoSb3through a combination of ab initio total-energy calculations and thermodynamics. Formation energies of void filling, Sb-substitution, and complex dual-site occupancy defects with different charge states, and their dependence on chemical potentials of species are considered. Our results show that Ga atoms predominantly form the dual-site 2GaVF-GaSbdefects and substitute for Sb only at very high Fermi levels or electron concentrations. Indium atoms, on the other hand, can play multiple roles in skutterudites, including filling in the crystalline voids, substituting for Sb atoms, or forming dual-site occupancy, among which the fully charge-compensated dual-site defects (2InVF-InSband 4InVF-2InSb) are dominant. The total defect concentrations are studied by using overall charge neutrality under the grand canonical ensemble. The concentration ratio of impurities at void-filling sites and that at Sb-substitution sites for Ga-doped CoSb3is very close to be 2∶1, while this value visibly deviates from 2∶1 for In-doped CoSb3. The 2∶1 ratio of Ga-doping in CoSb3causes low electron concentration (~2*1019cm-3) and makes the doped system a semiconductor. The underlying physics of the doping behavior for group 13 elements in CoSb3is also analyzed.
Key words:skutterudites; ab initio; thermoelectric materials; complex defects
中图分类号:TB34
文献标识码:A
文章编号:1674-3962 (2015)01-0041-09
DOI:10.7502/j.issn.1674-3962.2015.01.04
基金项目:国家自然科学基金资助项目(11204333, 11234012); 科技部“973”计划项目(2013CB632501)
收稿日期:2014-11-06
第一作者及通讯作者:席丽丽,女,1981年生,助理研究员,Email:lilyxi2006@mail.sic.ac.cn
