HPLC梯度洗脱法测定氧氟沙星有关物质的方法研究
2015-02-24曹晓云袁雯玮
张 琳,曹晓云,袁雯玮
(天津市药品检验所,天津 300070)
HPLC梯度洗脱法测定氧氟沙星有关物质的方法研究
张 琳,曹晓云,袁雯玮
(天津市药品检验所,天津 300070)
目的:建立HPLC梯度洗脱方法同时测定氧氟沙星中的杂质A和有关物质。方法:采用反相高效液相色谱法,色谱柱为Agilent XDB-C18(250 mm×4.6 mm,5 μm);以醋酸铵高氯酸钠溶液(取醋酸铵4.0 g和高氯酸钠7.0 g,加水1 300 ml使溶解,用磷酸调节pH值至2.2)-乙腈(85∶15)为流动相A,乙腈为流动相B,进行线性梯度洗脱;流速为1 ml/min;柱温为40 ℃;检测波长为238 nm和294 nm。结果:样品中各杂质峰及主峰的分离度和检测灵敏度均满足有关物质的测定要求。结论:方法灵敏、准确、专属性强,可同时测定氧氟沙星的杂质A和有关物质。
氧氟沙星,有关物质,杂质A,高效液相色谱法,梯度洗脱
氧氟沙星为喹诺酮类抗菌药,具有广谱抗菌作用,尤其对需氧革兰阴性杆菌抗菌活性高。氧氟沙星由日本第一制药公司在1980年研制成功,1985年4月获批准上市。在国内,氧氟沙星由浙江新昌制药厂和中国医学科学院医药生物技术研究所等单位首先仿制,于1992年6月被批准上市。
2010年之前《中国药典》、《欧洲药典》/《英国药典》和《美国药典》均以醋酸铵高氯酸钠溶液(取醋酸铵4.0 g和高氯酸钠7.0 g,加水1 300 ml使溶解,用磷酸调节pH值至2.2)-乙腈(85∶15)为流动相系统等度洗脱法检查氧氟沙星的有关物质, EP/BP在正文后列出了其7个已知杂质的结构[1,2],并采用薄层色谱法检查杂质A,国内尚未见相关的文献报道。本文参考《中国药典》2005年版[3]流动相系统,研究建立了测定氧氟沙星有关物质的HPLC梯度洗脱方法,使保留较强的杂质A及其他未知杂质得到有效洗脱,并确定了更为科学有效的分离度试验溶液。
本文建立的方法可在测定氧氟沙星有关物质的同时对杂质A进行定量检查,较国内外现有的同品种质量标准更有优势,已被《中国药典》2010年版收载。该方法较各国药典现有方法能更真实、准确地反映氧氟沙星中有关物质的情况,有助于发现产品的质量问题,可以更有效地控制生产工艺的稳定性,进而促进产品质量的提高。
1 仪器与试药
1.1 仪器 Agilent 1200高效液相色谱仪(DAD检测器)和岛津LC-20A高效液相色谱仪。
1.2 试药 醋酸铵、高氯酸钠和磷酸均为分析纯,乙腈为色谱纯。氧氟沙星对照品(批号130454-200604,HPLC纯度98.6%)、氧氟沙星杂质A、B、E对照品(杂质对照品仅供科研用)和环丙沙星对照品(批号130451-200302,HPLC纯度84.9%,供含量测定用)均由中国药品生物制品检定所提供。氧氟沙星(批号0709271、0710191、0712262、OF-M20050705、OF-M20080413和OF-M20080414)及制剂均为国内药厂生产。
2 方法与结果
2.1 色谱条件 色谱柱为Agilent XDB-C18(250 mm×4.6 mm,5 μm);以醋酸铵高氯酸钠溶液(取醋酸铵4.0 g和高氯酸钠7.0 g,加水1 300 ml使溶解,用磷酸调节pH值至2.2)-乙腈(85∶15)为流动相A,乙腈为流动相B,线性梯度洗脱,时间程序见表1。流速为1 ml/min;柱温为40 ℃;检测波长为238 nm和294 nm。

表1 梯度洗脱程序
2.2 溶液的制备
2.2.1 供试品溶液 取本品适量,用0.1 mol/L盐酸溶液制成每1 ml中约含1.2 mg的溶液。
2. 2.2 对照溶液 精密称取氧氟沙星对照品适量,加0.1 mol/L盐酸溶液溶解并稀释制成每1 ml中含2.4 μg的溶液。
2.2.3 杂质A对照品溶液 精密称取杂质A对照品约12 mg,置100 ml量瓶中,用6 mol/L氨溶液0.6 ml与水适量使溶解,并用水稀释至刻度,摇匀,精密量取2 ml,置100 ml量瓶中,加水稀释至刻度,摇匀,作为杂质A对照品溶液,用于杂质A定量。
2.3 系统适用性试验 称取氧氟沙星对照品、环丙沙星对照品和杂质E对照品各适量,用0.1 mol/L盐酸溶液溶解并稀释制成每1 ml含氧氟沙星1.2 mg、环丙沙星和杂质E各6 μg的混合溶液。取10 μl注入液相色谱仪,在 294 nm波长下记录色谱图,氧氟沙星峰的保留时间约为15 min,氧氟沙星峰与杂质E峰和环丙沙星峰间的分离度均应不小2.0,见图1。

1.杂质B 2.杂质E 3.氧氟沙星4.环丙沙星 5.杂质A 6.杂质1 7.杂质2
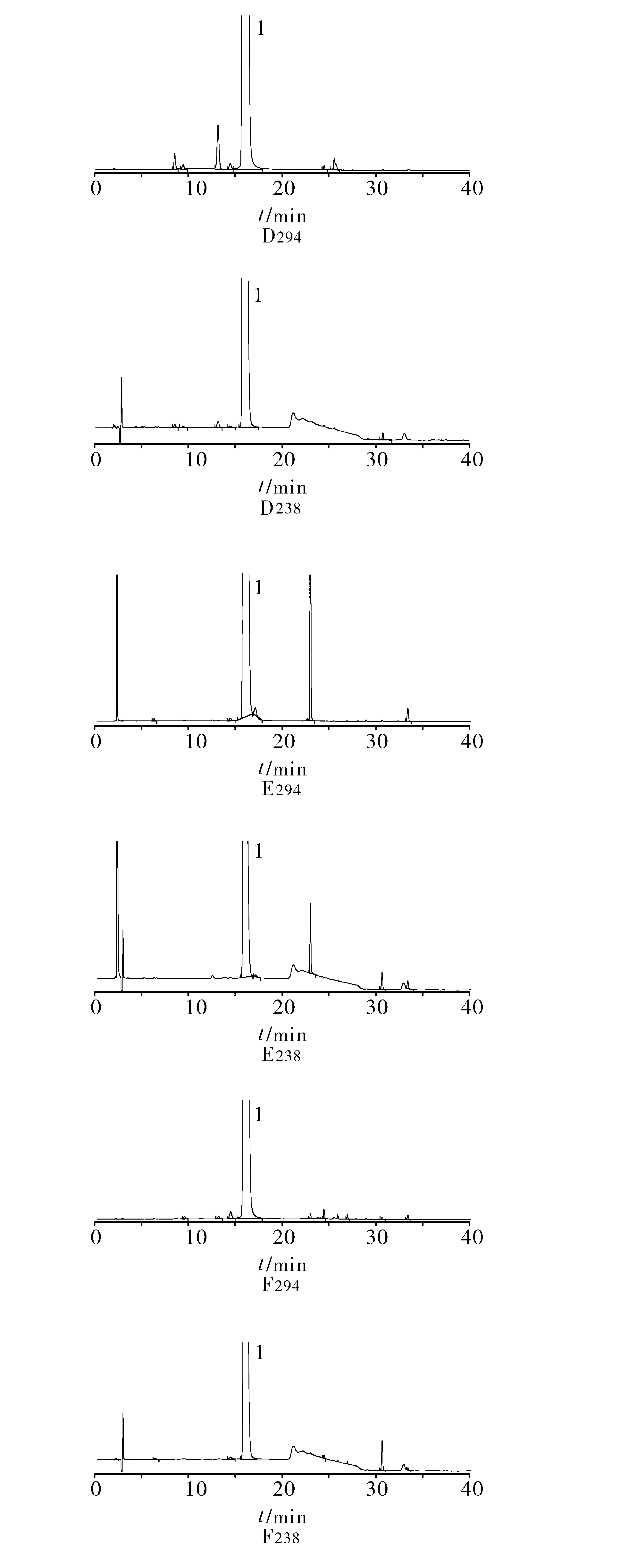
2.4 专属性试验 取氧氟沙星原料,分别经酸、碱、光、氧化和热破坏后,在238 nm和294 nm进样测试。结果表明,在各种破坏条件下所得的降解产物与主成分峰分离良好,降解产物峰之间也均能达到基线分离,该方法专属性好,结果见图2。
2.5 线性关系考查 取氧氟沙星和杂质A对照品各适量,分别制成每1 ml中约含0.06 mg的储备溶液,精密量取储备溶液各0.5、1、2、5和10 ml置50 ml量瓶中,制成系列浓度的溶液,进样测试,考查氧氟沙星(294 nm检测)和杂质A(238 nm检测)的线性关系。结果表明,在294 nm检测,氧氟沙星溶液浓度在0.6~30 μg/ml范围内,峰面积与浓度均呈良好的线性关系,回归方程为Y=5.557 8×104X+3.984 2×102(r=1.000 0);在238 nm检测,杂质A溶液浓度在0.6~30 μg/ml范围内,峰面积与浓度均呈良好的线性关系,回归方程为Y=5.528 9×104X+2.460 2×103(r=1.000 0)。
2. 6 检出限 在238 nm检测,杂质A最低检出浓度为0.048 μg/ml(相当于供试品溶液的0.004%);在294 nm检测,杂质B最低检出浓度为0.012 μg/ml(相当于供试品溶液的0.001%),杂质E最低检出浓度为0.024 μg/ml(相当于供试品溶液的0.002%),氧氟沙星最低检出浓度为0.024 μg/ml(相当于供试品溶液的0.002%)。
2. 7 溶液的稳定性 氧氟沙星供试品溶液室温条件下放置0、1、2、4和8 h时,分别进样测试,以峰面积计算,杂质A、最大单一杂质和总杂质的RSD分别为0.5%、1.2%和1.0%(n=5)。表明供试品溶液室温条件下放置8 h内基本稳定。
2. 8 供试品溶液浓度与有关物质测定结果的相关性 取氧氟沙星样品,分别制成浓度为0.96、1.2和1.44 mg/ml的供试品溶液和相应的0.2%的自身对照溶液。分别依法测定,以峰面积计算,杂质A、最大单一杂质和总杂质的RSD分别为1.5%、1.3%和0.8%。表明供试品溶液在0.96~1.44 mg/ml浓度范围内,有关物质测定结果与溶液浓度有良好的相关性。
2.9 方法耐用性 采用①Agilent XDB-C18(250 mm×4.6 mm,5 μm)、②Agilent ZORBAX SB-C18(250 mm×4.6mm 5μm)、③菲罗门 luna C18(250 mm×4.6 mm,5 μm)和④SGE C18(250 mm×4.6 mm,5 μm)4根不同色谱柱考查分离度测试溶液,主峰与杂质E峰和环丙沙星峰间的分离度均不低于2.0。采用条件①Agilent 1200高效液相色谱仪,Agilent XDB-C18(250 mm×4.6 mm,5 μm)色谱柱和条件②岛津LC-20A高效液相色谱仪,Agilent ZORBAX SB-C18(250 mm×4.6 mm,5 μm)同时考查相同2批样品的有关物质,测定结果基本一致。上述试验结果表明该方法耐用性良好。
2. 10 样品的测定结果 对2个生产厂家提供的6批氧氟沙星原料采用以上方法测定有关物质,结果见表2。
2. 11 有关物质限度的确定 本方法采用杂质对照品外标法对杂质A进行定量;对于除杂质A外其他杂质,采用不加校正因子的主成分自身对照法测定,与EP和BP一致。由于采用HPLC梯度洗脱法,本法较同品种其他质量标准多检出杂质A和一较大的未知杂质,根据样品的测定结果及EP对杂质A所规定的限度,本方法限度规定见表3。


1.氧氟沙星

表3 有关物质限度表
3 讨论
3.1 关于系统适用性试验 《欧洲药典》/《英国药典》及《中国药典》2005年版系统适用性溶液均限定氧氟沙星峰与杂质E峰(相对保留时间约为0.9)的分离度,未对主峰后紧邻杂质的分离加以控制。而图2供试品溶液色谱图中标示的未知杂质“杂质1”在部分氧氟沙星原料及制剂中均可检出,最高可达0.25%,因此对氧氟沙星与其后杂质的分离度加以限定是必要的。对不含杂质1的样品进行各种强制破坏后均未得到主峰后紧邻的杂质;以梯度洗脱系统考查环丙沙星、诺氟沙星等十几种同类物质,得到环丙沙星峰与氧氟沙星峰的相对保留时间约为1.1,与杂质1的保留时间最为接近。因此规定以氧氟沙星与杂质E(相对保留时间0.9)和环丙沙星(相对保留时间1.1)进行系统适用性分离度的试验,分离度均不低于2.0,可保证主峰与其前后相邻杂质峰有效分离。
3.2 检测波长的选择 《中国药典》2005年版、《欧洲药典》/《英国药典》均采用294 nm作为检测波长。经DAD试验结果表明,氧氟沙星和杂质A分别在294 nm和238 nm处有最大吸收,杂质E、杂质1和杂质2最大吸收波长均在294 nm附近;故本方法选择在238 nm波长处检查杂质A,在294 nm波长处检查其他杂质。
3.3 氧氟沙星制剂的测定 有关物质测定方法的关注点在于方法的灵敏度和专属性,即杂质被有效地检出。本文采用梯度洗脱的方法,不仅能将保留较强的杂质A洗脱,还可检出更多未知杂质,更能体现样品的真实情况;采用294 nm和238 nm双波长检测还可同时对杂质A进行定量,较薄层色谱法更为准确。采用本文方法分析不同企业生产的氧氟沙星片、胶囊、注射液和滴眼液等制剂,辅料均无干扰,因此本方法可同时用于氧氟沙星系列制剂的有关物质(包括杂质A)测定。
1 European Pharmacopoeia 6.0[S].2007:2541-2542
2 The British Pharmacopoeia 2009[S]. 2008:1578-1579
3 中国药典[S].二部.2005:605
Determination of related substances of ofloxacin by gradient elution HPLC
Zhang Lin,Cao Xiaoyun,Yuan Wenwei
(Tianjin Institute for Drug Control,Tianjin 300070)
Objective:To establish a method for the determination of impurity A and related substances of ofloxacin by gradient elution HPLC. Methods :The separation was performed on a Agilent XDB-C18(250 mm×4.6 mm ,5 μm) column. Ammonium acetate and sodium perchlorate solution(dissolve 4.0 g of ammonium acetate and 7.0g of sodium perchlorate in 1 300 ml of water, adjusted to pH 2.2 by phosphoric acid) -acetonitrile(85∶15) as mobile phase A, acetonitrile as mobile phase B.The flow rate was 1 ml/min, the column temperature was 40 ℃, detective wavelength was 238 nm and 294 nm. Results: The resolution and sensitivity of related substances were acceptable. Conclustion: The method is sensitive,accurate and specific and can be used for the determination of impurity A and the related substances of ofloxacin.
ofloxacin,related substances,impurity A,HPLC,gradient elution
2014-04-21
R927.11
A
1006-5687(2015)01-0005-05
