Rho激酶介导醛固酮致人近端肾小管上皮细胞间充质转分化
2015-02-23孙广萍李德天
孙广萍,朱 凯,李德天*
Rho激酶介导醛固酮致人近端肾小管上皮细胞间充质转分化
孙广萍1,朱 凯2,李德天1*
目的 探讨醛固酮(Aldosterone,ALD)能否诱导人近端肾小管上皮细胞(HK2细胞)发生上皮间充质转分化(Epithelial-mesenchymal transition,EMT)以及Rho激酶在此过程中的作用。方法 用无血清DMEM/F-12培养液同步化培养HK2细胞24 h后,将HK2细胞分为对照组、ALD组、ALD+螺内酯(盐皮质激素受体拮抗剂)组,ALD+Y-27632(Rho激酶抑制剂)组,观察各组间E-cadherin、α-SMA 蛋白(Western blot法)及mRNA(real-time PCR法)表达,以及各组间磷酸化MYPT-1的表达(Western blot法)。结果 终浓度为10-7M的ALD作用下,随作用时间延长,HK2细胞 E-cadherin蛋白及mRNA表达逐渐减少,α-SMA蛋白及mRNA表达逐渐增多,提示HK2细胞发生了EMT。与对照组相比,ALD组E-cadherin蛋白及mRNA表达明显减少(P<0.05),α-SMA蛋白及mRNA表达显著增多(P<0.05),伴有磷酸化MYPT-1表达增多;而螺内酯及Y-27632均明显减轻了上述改变,与醛固酮组比较,差异有统计学意义(P<0.05)。结论 Rho激酶活化介导了醛固酮诱导的HK2细胞发生EMT。
醛固酮;EMT;Rho激酶;HK2细胞
0 引言
肾小管间质纤维化是肾功能进行性进展至终末期肾病的共同途径,其比肾小球病变更易引起肾功能进展亦成为共识。近年来研究认为,多种因素可以诱导肾小管上皮细胞发生上皮间充质转分化(Epithelial mesenchymal transition,EMT)[1],使肾小管上皮细胞首先转变为肌成纤维细胞,再转变为纤维细胞,是促进肾间质纤维化的重要途径之一。
通常情况下,醛固酮(ALD)作为盐皮质激素调节机体的水盐代谢。但近年来发现,ALD有促进器官纤维化的作用,除心脏、血管、脑外,在肾脏的系膜细胞、足细胞、肾小管上皮细胞以及间质成纤维细胞上都存在盐皮质激素受体(Mineralosteroid receptor,MR)[2]。在ALD灌注大鼠[3],不仅存在严重的肾小球损伤,还表现明显的肾小管间质纤维化,伴随有肾小管上皮细胞表达α-SMA增多,表明ALD可能有诱导肾小管上皮细胞发生EMT的作用。
Rho激酶是小G蛋白Rho的一个效应因子,激活后调节肌动、肌球蛋白交联增加,促进应力纤维和粘附斑的形成,调节肌动蛋白细胞骨架的聚合,从而介导包括平滑肌收缩、细胞与基质及细胞与细胞间粘附、细胞迁移、增生、分化等与肾脏损伤相关的细胞功能[4-5]。近年来许多体内体外研究提示,Rho/Rho激酶通路参与调节肾小管上皮细胞发生EMT,从而引起肾间质纤维化[6-7]。
笔者研究发现,在ALD作用下,大鼠出现大量蛋白尿及肾脏纤维化的同时,伴有Rho激酶活性增强,肾小管上皮细胞表达α-SMA增多,应用Rho激酶抑制剂fasudil抑制Rho激酶活性,虽无明显降压作用,却明显减轻了肾间质纤维化,肾小管上皮细胞表达α-SMA减少,提示ALD可能通过激活Rho激酶诱导肾小管上皮细胞发生EMT[3]。本研究旨在利用人近端肾小管上皮细胞HK2细胞系,体外予ALD刺激,探讨ALD能否通过Rho激酶途径诱导肾小管上皮细胞发生EMT。
1 材料与方法
1.1 实验材料
1.1.1 人HK2细胞系 购于美国ATCC细胞平台,在6孔板上接种HK2细胞,用10% FBS的DMEM/F-12培养液、置CO2培养箱(5% CO2,37 ℃)培养,待细胞生长达到70%融合,换用无血清DMEM/F-12培养液,置5%CO2培养箱中培养。静置24 h后细胞同步化进行条件干预。
1.1.2 主要试剂 DMEM/F-12培养液、小牛血清、青霉素、链霉素、胰蛋白酶购于美国Hyclone 公司,ALD粉剂(Sigma)、螺内酯(Spirolactone,Spi)购于Sigma;Y-27632(RB),兔抗人E-cadherin抗体,鼠抗人α-SMA抗体,鼠抗人GAPDH抗体购自北京中杉生物技术有限公司;鼠抗人磷酸化MTYPT-1抗体(Santa Cruz)。RIPA全蛋白裂解液购自碧云天公司,磷酸化/去磷酸化保护剂购自凯基公司。TRIzol购自美国Invitrogen公司,E-cadherin、α-SMA及GAPTH引物由美国Invitrogen公司合成;逆转录试剂盒及定量试剂盒购自日本 TAKARA公司。
1.2 实验方法
1.2.1 细胞处理 将5~6代的HK2传代于6孔细胞培养板中培养,待细胞融合率达70%,换无血清DMEM/F-12培养液培养24 h,加入终浓度为10-7M的ALD,检测不同时间点(0、12、24、48 h)HK2细胞α-SMA、E-cadherin蛋白及mRNA表达。
1.2.2 细胞分组 对照组(C组):无血清 DMEM/F-12 培养液。ALD组:ALD 终浓度10-7M。Spi组:在ALD干预前,螺内酯预孵6 h,终浓度10-7M。Y-27632组:在ALD干预前,Y-27632预孵1 h,终浓度10-6M。各组均设3复孔。
1.2.3 Western blot法检测E-cadherin、α-SMA、磷酸化MYPT-1 具体如下:蛋白提取全程冰上操作,PBS冲洗6孔板3~4遍,为检测E-cadherin、α-SMA、直接加入裂解液50 μL/孔[裂解液配制:碧云天RIPA全蛋白裂解液1 mL中加入磷酸酯酶抑制剂(PMSF)10 μL]。为检测磷酸化MYPT-1蛋白,直接加入裂解液50 μL/孔(裂解液配制:碧云天RIPA全蛋白裂解液1 mL中加入磷酸化/去磷酸化磷酸酶保护剂10 μL)。之后冰上裂解15 min,刮取裂解物并收集,4°离心25 min,取上清液,收集上清液,考马斯亮蓝法测蛋白浓度,然后应用1倍sample buffer进行蛋白变性,取400 μg蛋白上样行SDS凝胶电泳。硝酸纤维素膜转膜后,膜置入TBS缓冲液中1 min,之后用含10%脱脂奶粉的TBS液封闭非特异抗体90 min,取出PVDF膜,用0.l%TBS-tween缓冲液,洗3次,每次5 min,然后分别加入TPBs-tween缓冲液稀释的兔抗人E-cadherin抗体(1∶200)、鼠抗人α-SMA抗体(1∶200)、鼠抗人磷酸化MYPT-1抗体(1∶400)室温孵育过夜9 h左右,用0.1%TPBS-tween缓冲液洗3次,每次5 min,然后分别加入用TPBS-tween缓冲液稀释的辣根酶标记的羊抗兔二抗(1∶2 000)、兔抗鼠二抗(1∶2 000)抗体,室温孵育2 h后用0.1%TPBS-tween缓冲液洗3次,每次10 min,然后加入ECL或ECLPluS暗室曝光,直到得到清晰条带为止。
用 NIH1.61图像分析软件分别测定目的蛋白条带密度并进行定量分析。以GAPDH为内参照,用目的蛋白与GAPDH条带密度的比值表示每个样本的相对蛋白表达水平。
1.2.4 real-time PCR法检测E-cadherin、α-SMA mRNA 表达水平 美国Medline国立图书馆基因库检索基因,PCR引物设计在primer 5.0引物设计辅助软件上进行,美国Invitrogen生物技术有限公司合成。提取肾组织总RNA,取2 μg RNA进行逆转录合成cDNA,然后采用 SYBR®Premix Ex TaqTMⅡ(Takara)基因表达测定试剂盒在ABI
Prism 7 000序列检测系统(Applied Biosystems,Foster City,CA)进行real-time PCR 分析。变性95 ℃ 30 s,扩增95 ℃ 5 s,60 ℃ 30 s,45个循环。以GAPDH作为内参照,分别计算目的基因与GAPDH 的比值。各靶基因引物序列见表1。

表1 PCR引物序列

2 结果
2.1 ALD作用不同时间对E-cadherin、α-SMA蛋白及mRNA表达的影响 研究表明,ALD浓度为10-7M,更接近CKD 3期患者的ALD浓度。所以选择10-7mol/L ALD作为工作浓度。分别给予不同的时间干预(0、12、24、48 h)。HK2细胞在ALD作用下,随作用时间的延长,E-cadherin蛋白及mRNA表达明显减少,α-SMA蛋白及mRNA表达明显增加(见图1),提示呈时间依赖性。本实验选择作用时间48 h。
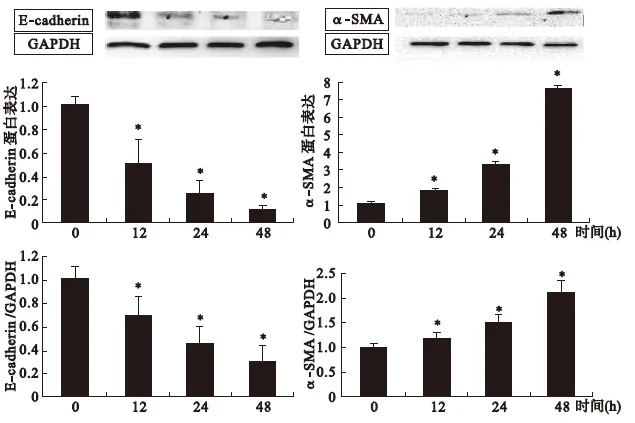
图1 不同时间点10-7M ALD作用下E-cadherin、α-SMA蛋白及mRNA的表达
2.2 不同组间E-cadherin、α-SMA蛋白及mRNA表达 与对照组比较,10-7M ALD作用下,E-cadherin蛋白及mRNA表达明显减少,α-SMA蛋白及mRNA表达明显增加,差异有统计学意义(P<0.05),说明醛固酮诱导HK2细胞发生EMT;与醛固酮组比较,螺内酯及Y-27632组均能显著减少E-cadherin、α-SMA蛋白及mRNA表达变化,差异有统计学意义(P<0.05)。见图2。
2.3 不同组间磷酸化MYPT-1蛋白的表达 MYPT-1是Rho激酶的作用底物,其磷酸化水平代表Rho激酶的活性。与对照组比较,ALD组磷酸化MYPT-1表达明显增加,差异有统计学意义(P<0.05)。与醛固酮组比较,螺内酯及Y-27632组细胞磷酸化MYPT-1表达明显减少,差异有统计学意义(P<0.05)。见图3。
3 讨论
ALD是调节机体水盐代谢的主要皮质激素,既往认为其生物学作用主要为促进肾脏远曲小管和集合管重吸收钠和水,排出钾。但许多研究表明醛固酮与肾脏纤维化关系密切,近年来发现,慢性肾脏病患者肾脏局部肾素-血管紧张素-醛固酮系统异常活化,ACEI/ARB长期治疗可在一定程度上延缓肾功能的进展。但是在某些患者会出现“ALD逃逸”现象,影响ACEI/ARB疗效。而应用MR拮抗剂螺内酯或依普利酮能进一步减少蛋白尿,保护肾小管间质,延缓肾功能进展,由此表明ALD是一个独立的CKD进展的危险因素[8]。
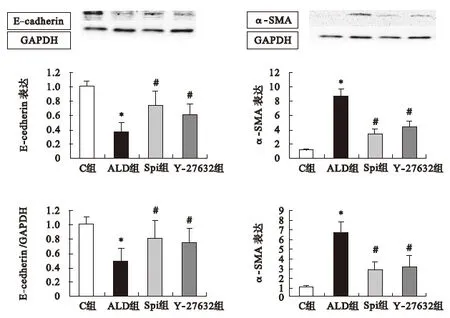
图2 不同组间E-cadherin、α-SMA蛋白及mRNA表达

图3 不同组间磷酸化MYPT-1蛋白的表达
本实验结果显示,体外培养人HK2细胞,在浓度10-7M的ALD作用下,随着作用时间的延长,肾小管上皮细胞标志物E-cadherin mRNA和蛋白表达均明显减少,而肌成纤维细胞标志物α-SMA mRNA和蛋白表达明显增高,且呈时间依赖,说明ALD能在体外诱导HK2细胞发生EMT。此结果与既往的研究结果一致[9-10],同时也进一步证明了之前的在体研究应用ALD灌注大鼠,大鼠的肾小管上皮细胞大量表达α-SMA,导致肾小管间质纤维化的研究结果。而应用MR拮抗剂 Spi,能明显抑制ALD作用后HK2细胞E-cadherin、α-SMA蛋白及mRNA表达的变化,说明ALD诱导EMT可能通过与HK2细胞上的盐皮质激素受体起作用。可能的机制为ALD与胞质内的MR 结成“ALD-受体”复合物,并移入胞核与反应元件结合,激活或抑制目的基因转录,减少上皮细胞极性标志蛋白E-cadherin的表达,上调转分化标志蛋白α-SMA的表达,促进EMT的发生。Spi作为 MR拮抗剂可与MR竞争结合,减少α-SMA的表达,上调E-cadherin 的表达,抑制EMT的发生,起到肾脏保护作用,但其确切的机制有待进一步探讨。
Rho/Rho激酶通路存在于大多数细胞中,是参与细胞有丝分裂、粘附、细胞骨架调整、细胞收缩、肿瘤细胞浸润等一系列细胞生命现象的重要酶。Rho激酶的经典底物是MYPT-1,也是最早发现的天然底物。Rho激酶活化后,催化其肌球蛋白结合亚单位(MYPT-1)磷酸化,使肌球蛋白磷酸酶失活;失活的肌球蛋白磷酸酶不能将肌球蛋白轻链去磷酸化,使得胞浆内磷酸化的肌球蛋白轻链水平上升,肌动、肌球蛋白交联增加,促进应力纤维和粘附斑的形成,调节肌动蛋白细胞骨架F-actin聚合。F-actin是参与细胞运动的主要蛋白质分子,因而也认为Rho激酶是调节细胞运动的重要激酶之一。很可能Rho激酶激活通过改变HK2细胞骨架蛋白F-actin 的重排,因而获得肌成纤维细胞的表型而促进 EMT的发生[11]。目前无论糖尿病肾病或肾小管间质纤维化的动物模型都显示,Rho/Rho激酶通路激活参与肾小管间质纤维化[12-13],而且细胞学研究也证明,Rho激酶不仅介导系膜细胞发生EMT,而且也介导肾小管上皮细胞发生EMT[10,14]。因此Rho/Rho激酶可能是导致EMT并最终至脏器纤维化的重要通路。笔者之前研究发现,在大鼠持续泵入ALD后,大鼠肾小管上皮细胞表达α-SMA增多,同时伴有Rho激酶的激活。本研究亦发现,在ALD诱导HK2细胞发生EMT的同时,伴有Rho激酶活性的标志物磷酸化MYPT-1蛋白表达增多。Spi及Rho激酶抑制剂Y-27632能减少磷酸化MYPT-1表达,同时HK2细胞EMT改变减轻。这提示Rho激酶激活参与ALD诱导的HK2细胞的EMT,与Wei等[10]结果一致,也进一步证实了先期研究Rho/Rho激酶参与介导醛固酮肾脏损伤的研究成果,但是也有报道,ALD很可能通过激活HK2细胞线粒体的氧化反应参与HK2细胞EMT[9-10]。因此,进一步研究希望通过转染Rho激酶siRNA 敲除或减少HK2细胞Rho激酶表达,观察ALD能否诱导细胞发生EMT,为ALD诱导HK2细胞发生EMT的机制研究提供新的靶点和依据。
[1] Barnes JL,Glass JW.Renal interstitial fibrosis: a critical evaluation of the origin of myofibroblasts[J].Contribu Nephrolol,2011,169:73-93.
[2] Brown NJ.Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis[J].Nat REV Nephrol,2013,9(8):459-469.
[3] Sun GP,Kohno M,Guo P,et al.Involvements of Rho-kinase and TGF-beta pathways in aldosterone-induced renal injury[J].J Am Soc Nephrol 2006,17: 2193-2201.
[4] Moriyama T,Nagatoya K.The Rho-ROCK system as a new therapeutic target for preventing interstitial fibrosis[J].Drug News Perspect,2004,17:29-34.
[5] Jan Danser AH.Blood pressure-independent effects of Rho-kinase inhibitors in the kidney[J].J Hypertens,2004,22:1675-1677.
[6] Liu M,Gu M,Wu Y,et al.Therapeutic effect of Y-27632 on chronic allograft nephropathy in rats[J].J Surg Res,2009,157(1):e117-e127.
[7] Zhang K,Zhang H,Xiang H,et al.TGF-β1 induces the dissolution of tight junctions in human renal proximal tubular cells: role of the RhoA/ROCK signaling pathway[J].Int J Mol Med,2013,32(2):464-468.
[8] Mavrakanas TA,Gariani K,Martin PY.Mineralocorticoid receptor blockade in addition to angiotensin converting enzyme inhibitor or angiotensin II receptor blocker treatment: an emerging paradigm in diabetic nephropathy: a systematic review[J].Eur J Intern Med,2014,25(2):173-176.
[9] Zhang AH,Jia ZJ,Guo XH,et al.Aldosterone induces epithelial-mesenchymal transition via ROS of mitochondrial origin[J].Am J Physiol Renal Physiol,2007,293:F723-F731.
[10]Wei JL,Li ZR,Ma CY,et al.Rho-kinase pathway is likely responsible for the probrotic actions of aldosterone in renal epithelial cells via inducing epithelial-mesenchymal transition and extracellularmatrix excretion[J].Cell Biol Int,2013,37:725-730.
[11]高文波,魏军军,翁国斌,等.移植肾间质纤维化和小管萎缩与肾小管上皮间充质转化的关系[J].实用医学杂志,2013,29(7):1083-1086.
[12]Tokuyama H,Wakino S,Hara Y,et al.Role of mineralocorticoid receptor/Rho/Rho-kinase pathway in obesity-related renal injury[J].International Journal of Obesity,2012,36(8):1062-1071.
[13]Nagatoya K,Moriyama T,Kawada N,et al.Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction[J].Kidney Int,2002,61(5):1684-1695.
[14]Diah S,Zhang GX,Nagai Y,et al.Aldosterone induces myofibroblastic transdifferentiation and collagen gene expression through the Rho-kinase dependent signaling pathway in rat mesangial cells[J].Exp Cell Res,2008,314(20):3654-3662.
Rho-kinase involved in the EMT induced by aldosterone in human proximal tubular epithelial cells
SUN Guang-ping1,ZHU Kai2,LI De-tian1*
(1.Department of Nephrology,Shengjing Hospital of China Medical University,Shenyang 110004,China ;2.Department of Nephrology,General Hospital of Fushun Minining Bureau,Fushun 113000,China)
Objective To investigate whether aldosterone (ALD) can induce EMT in the human proximal tubular epithelial cell- HK2cells and the role of Rho-kinase in the process.Methods HK2cells were simultaneously cultured with serum-free DMEM/F-12 for 24 h.Then the cells were divided into the following four groups: control group(only DMEM/F-12),ALD group (ALD 10-7M),ALD+spironolactone group (ALD 10-7M and preincubation with spironolactone 10-7M) and ALD+Y27632 group (ALD 10-7M and preincubation with Y-27632 10-6M).The protein and mRNA expressions of E-cadherin and α-SMA,both with the phosphoralation of MYPT-1 were compared among the groups by real-time PCR and/or Western blot.Results Stimulation with aldosterone significantly decreased the expressions of E-cadherin protein and mRNA and increased the expressions of α-SMA protein and mRNA,which suggested EMT in the HK2cells.The phosphorated-MYPT-1 protein,marker of Rho-kinase activity simultaneously increased markedly.Preincubation with spironolactone(10-7M) or a Rho-kinase inhibitor Y-27632 (10-6M)attenuated the aldosterone-induced increase in MYPT-1 phosphorylation,accompanied by attenuation of EMT.Conclusion Activation of Rho-kinase is involved in the EMT of HK2cells induced by ALD stimulation.
Aldosterone;EMT;Rho-kinase;HK2cells
2014-11-31
1.中国医科大学附属盛京医院肾内科,沈阳
110004;2.抚顺矿务局总医院肾内科,抚顺 113000
辽宁省博士启动基金(20101143)
10.14053/j.cnki.ppcr.201508009
*通信作者
