弥漫性轴索损伤后胶质反应及生化标记物的研究进展
2015-02-23宋锦宁赵永林
宋锦宁,赵永林
(西安交通大学医学院第一附属医院神经外科,陕西西安 710061)
◇专家述评◇
弥漫性轴索损伤后胶质反应及生化标记物的研究进展
宋锦宁,赵永林
(西安交通大学医学院第一附属医院神经外科,陕西西安 710061)

弥漫性轴索损伤(diffuse axonal injury, DAI)后胶质反应与继发性轴索损伤、轴突再生及患者的预后密切相关,而且DAI后一些特异性生化标志物可以发生明显的变化。目前的研究表明,DAI后胶质细胞既能通过胶质瘢痕形成、炎症反应、髓鞘丢失等多种机制参与DAI后继发性损伤的发生,又能通过释放神经营养因子、清除细胞碎片、阻止病损蔓延等促进轴突修复和再生,胶质细胞之间、胶质细胞与神经元之间的相互作用是最终导致DAI后神经元死亡以及轴突继发性断裂的主要原因;DAI后β淀粉样蛋白、神经微丝和微管相关蛋白等特异性生化指标能更早期、更准确地显示DAI的发生与发展,将弥补影像学及临床特征诊断的不足。这些研究不但为DAI后神经保护及轴突再生提供治疗靶点,也为进一步研究并寻找新的DAI相关生化标记物提供理论依据。进一步阐明DAI后胶质细胞激活、胶质细胞与神经元相互作用、胶质细胞之间相互作用的相关分子机制及其复杂的病理生理过程,以及深入揭示DAI相关生化标志物的意义,对减轻DAI后脑损伤、促进轴突再生、提高DAI诊断及治疗水平具有重要意义。
颅脑损伤;弥漫性轴索损伤;胶质反应;生化标记物
弥漫性轴索损伤(diffuse axonal injury, DAI)是一种特殊类型的颅脑损伤,以广泛神经轴索断裂为特征,常导致患者高致残率和死亡率。对DAI后神经元进行性的病理生理变化及相应治疗策略的研究发现,一些以神经元为治疗靶点的药物,如钙离子拮抗剂、轴膜保护及修复药物、钙依赖的蛋白水解酶抑制剂等,并没有明显改善DAI患者的预后。这一结果提示,DAI后脑损伤发生的源头并不仅仅在神经元,越来越多的证据显示胶质细胞在DAI后神经轴索的病理变化中也起重要作用。DAI后小胶质细胞、星形胶质细胞由静态迅速激活,这一过程称为“反应性胶质化(reactive gliosis)”;而少突胶质细胞在受到刺激后死亡、丢失,其前体细胞开始增生、分化。研究者把DAI后胶质细胞一系列形态与功能的变化过程统称为“胶质反应(glial response)”[1-2]。近年来除了深入研究DAI后神经元继发性损伤的机制,探索胶质细胞的病理生理与分子变化也将为治疗DAI提供新方向。
目前,临床上仍没有诊断DAI的特异性手段,无论是CT还是MRI,在临床上均有一定的局限性,患者病情常得不到正确评估,延误了最佳治疗时间。故此,探索可用于诊断DAI的生化分子标记物对提高诊断率、及时了解病情变化、评估治疗效果和预后显得十分迫切。近年来,通过检测血清或脑脊液(cerebrospinal fluid, CSF)中DAI特异性生化指标来判断DAI病情严重程度、评估预后、寻找更佳的治疗方法及调整治疗方案等成为临床医生关注的热点。目前发现的一些分子,如NF、C-tau、SBDP、MBP、β-APP、FE65 mRNA、NSE等,在DAI后均特异性的表达,这些分子可作为DAI后特异性生化标记物。除了继续探索用于诊断和判断DAI预后的特异性生化指标,进一步确定这些标志物在CSF和血液中的时间-浓度变化规律及其诊断DAI阈浓度的大小,将使其在临床的应用前景更加广阔。
1 DAI后胶质细胞的病理生理变化
1.1 星形胶质细胞 星形胶质细胞(astrocyte, AST)来源于神经上皮,具有多个细小分枝,遍布神经系统。生理状态下,AST分化效率很低,对神经回路起营养支持、隔离、绝缘作用,调控突触释放神经递质;参与血脑屏障(blood brain barrier, BBB)的组成,调节血流量和呼吸,维持水、离子和pH的平衡,转运营养物透过BBB。生理状态下,AST内吞噬相关受体及其下游分子通路表达较高,表明AST有较强的吞噬能力,AST还是细胞外基质蛋白和黏附分子的主要来源,调控神经元的形态、存活、迁移、分化[3-4]。
DAI后成熟AST为应对损伤,出现以细胞增殖和细胞肥大为特征的反应性胶质化,也称AST激活或活化[5]。引起胶质化的刺激源仍未确定,但已有研究认为退化的、断裂的轴突、死亡的胞体或突触、BBB破坏后血清来源的分子和炎症相关细胞因子都可能是刺激AST反应性胶质化的物质[6]。这些细胞因子包括白介素1β(interleukin-1β, IL-1β)、肿瘤坏死因子α (tumor necrosis factor-α, TNF-α)和转化生长因子β1(transforming growth factor-β1, TGF-β1)等,AST表达这些因子的受体与之结合,导致AST激活[7]。
AST活化后,细胞内中间纤维丝胶质纤维酸性蛋白(glial fibrillary acidic protein, GFAP)和波形蛋白vimentin表达上调,AST胞体肿大,胞质混浊,核固缩或溶解,突起增粗、延长,并呈进行性改变,当损伤区的溃变产物被AST及其他吞噬细胞吞噬清除后,AST以其突起充填空隙,形成致密的胶质瘢痕[8]。通常认为损伤部位的反应性AST系从远处迁移而至,但也有研究者认为系损伤区域周围的AST增生所致[9]。
DAI后,AST活化、增殖,围绕兴奋性氨基酸、活性氧簇和自由基等毒素形成一道高密度的胶质瘢痕,阻断病损区域的坏死、感染及炎性细胞浸润,保护其他未受损的脑实质。AST内水通道蛋白4(aquaporin4, AQP4)的转录和表达会增加,引起细胞源性的脑水肿。当BBB受损引起血管源性的脑水肿时,AST会减少AQP4的转录和表达以减轻细胞源性的水肿。DAI后突触发生区域的AST会释放基质金属蛋白酶,重构损伤的细胞外基质蛋白,并参与清除坏死的细胞碎片,为突触发生和成熟提供适宜的环境[10]。反应性AST还能表达合成一些神经营养因子,如胰岛素样生长因子1和表皮生长因子等,能减少DAI所致的迟发性神经元死亡,但脑组织内营养因子的时空浓度有待进一步确认[11]。AST在神经损伤后能有效地聚集和吞噬死亡的神经元细胞,避免旁杀伤现象的发生,防止正常细胞凋亡,减轻神经损伤。
由于病损的持续存在,AST可能持续激活,过度活化。过度活化的AST可持续释放IL-1β、TNF-α等炎性细胞因子、氧自由基和细胞毒性物质,引起脑组织炎症反应和氧化应激反应,直接或间接诱发神经元死亡。过度增生的AST不仅形成胶质瘢痕,抑制神经再生,AST重构细胞外基质的作用也被AST释放的硫酸软骨素蛋白多糖(chondroitin sulfate proteoglycans, CSPGs)掩盖,这一蛋白可抑制轴突的生长[6]。因此,过度活化的AST对神经元轴突和髓鞘的再生构成结构和功能上的障碍,从而影响神经组织的修复。此外,DAI后活化的AST清除细胞碎片的能力降低,一方面可能因为活化后其吞噬能力降低,另一方面可能因为髓鞘碎片没有正确表达吞噬相关信号。AST介导的髓鞘清除对轴突生长及其长期功能恢复具有重要的作用,其具体机制仍需深入研究。
DAI后AST反应性活化包括生物分子表达、进行性的细胞肥大、增生和瘢痕形成等,且随着病损的性质和严重程度呈现一个连续的、有梯度的和进行性的改变。AST的活化对神经元有促损伤和促修复双重作用;但是目前对AST在DAI后扮演角色的时程缺少长时、系统、大量的观察和评价,保护和损伤作用是同时发生,还是呈现渐强或渐弱的趋势?在损伤的不同阶段AST是起损伤或修复作用仍不明确;抑制AST活化对神经元是有利亦或有害也未知[12]。
1.2 小胶质细胞 小胶质细胞(microglia, MG)被认为是中枢神经系统内(central nervous system, CNS)广泛分布的巨噬细胞,不断迁移并监测周围的微环境,参与神经系统的免疫应答,也参与轴突再生。生理状态下,MG处于静息状态,呈多级分枝状,胞体小,突起较细;DAI后损伤的轴索或升高的β淀粉样前体蛋白直接或间接激活了损伤部位及小血管周围的MG,MG迅速增殖、聚集成簇[13]。激活早期,MG突起变长变粗,呈高分枝状,染色深;晚期,突起变短,变成圆形、阿米巴状或杆状。伤后M1型巨噬细胞/MG的数量明显升高,持续至14 d,M2型在伤后5 d达到高峰,随后快速下降。M1型MG的数量变化趋势与DAI损伤程度明显一致,MG由M2向M1的表型转换可能也参与轴索损伤[14]。
轴索损伤后MG迁移至损伤或死亡细胞周围,细胞内吞噬相关的膜受体表达也升高,吞噬细胞碎屑和溃变的髓鞘,为神经元提供有利微环境,促进组织修复;MG也能吞噬损伤的神经细胞,直接或间接地引起神经元死亡[9]。MG激活后也可释放神经生长因子,脑源性神经营养因子和神经营养素等因子,促进神经元的存活和轴突再生;产生抗氧化酶清除自由基,减轻氧化应激,保护神经元;MG也可通过摄取细胞外谷氨酸,减轻谷氨酸对神经元的兴奋性毒性[15]。
另一方面,MG持续激活,产生大量兴奋性神经毒性氨基酸(如谷氨酸、天门冬氨酸等)和活性氧、活性氮中间体,介导神经元损伤。此外,DAI后活化的MG高度表达多种促炎细胞因子(IL-1α、IL-1β、IL-6、TNF-α等),介导炎症反应,微环境中大量的炎性细胞因子、趋化因子和氧自由基的释放反过来激活更多的MG活化,从而形成恶性循环[16]。随着进一步研究发现,人MG反应活性的高峰在伤后3月,若干年后才逐步恢复至正常水平。这可能由于DAI后脑急性反应促发MG的正反馈循环,直接影响神经元轴突或突触的功能和结构,介导伤后神经退行性病变的发生,而抑制MG的激活可改善DAI的长期预后。
DAI后MG的反应及释放的细胞因子的角色仍有争议,既可能起神经保护作用,又可能导致神经退行性病变。但具体MG是如何被激活的,它在神经轴索损伤“二次打击”的病理生理过程中起着怎样的作用还有待于进一步探讨。
1.3 少突胶质细胞 少突胶质细胞(oligodendrocyte)也起源于神经上皮,产生包绕神经元轴突的髓鞘质,少突胶质细胞及其前体细胞(oligodendrocyte precursor cells, OPCs)广泛分布在CNS的灰质及白质内。为维持髓鞘化轴突的结构和功能,少突胶质细胞的代谢率极高,消耗大量的氧气和ATP,因此对组织损伤也极敏感。正常情况下,与其他胶质细胞相比,少突胶质细胞介导的吞噬作用较弱,只能吸收较少髓鞘碎片[17]。
DAI后的缺血再灌注损伤使少突胶质细胞对氧化应激变得敏感;继发的缺氧及代谢障碍、线粒体损伤及诱发的氧化应激可引起少突胶质细胞坏死、凋亡及自噬,导致轴突脱髓鞘改变,轴突传导障碍,最终导致轴突死亡。谷氨酸和其他神经递质触发的钙离子信号在少突胶质细胞生长和和损伤的过程中起重要作用[18]。DAI后谷氨酸代谢异常、钙离子信号紊乱均能对少突胶质细胞的生理活动造成影响。静息状态的少突胶质细胞并不参加髓鞘再生[19]。DAI发生后,OPCs被激活、增殖,逐渐成熟,迁移到损伤区域,并向形成髓鞘的少突胶质细胞分化,促进轴突再生。少突胶质细胞也可产生胶质细胞源性神经营养因子、脑源性神经营养因子及胰岛素样生长因子-1,促进OPCs的成熟与分化。损伤范围和程度过大时,可导致永久的脱髓鞘改变,导致轴索功能缺陷和不可逆性变性。揭示DAI后少突胶质细胞支持轴突存活的机制也是当前研究DAI治疗策略的热点。
DAI发生后,各种静息状态的胶质细胞快速激活,处于反应性胶质化状态,MG的激活和持续的AST活化均涉及多种炎症介质的产生与释放,胶质细胞激活后反过来刺激周围的胶质细胞和神经元[20]。例如,研究证实AST释放的趋化因子CXCL 12可促进谷氨酸的释放,并产生少量TNF-α,这一系列刺激引发MG大量释放TNF-α[21]。高浓度TNF-α的存在使AST清除谷氨酸的能力受损,进而导致兴奋毒性对神经元造成损失。AST也可释放抗炎因子IL-10,并通过STAT-3通路抑制MG释放TGF-β,促进OPCs的成熟[22]。胶质细胞的激活使神经-胶质之间、胶质-胶质之间的交互影响变得活跃,既可引起突触连接的功能障碍,神经递质稳态失衡,又可能导致轴索变性和神经元死亡。揭示胶质-胶质/神经-胶质复杂网络间的相互作用及机制是一项十分艰巨的任务,也是目前研究的瓶颈所在。
目前,关于DAI后胶质反应的研究虽然取得了一定的进展,而实际上这些胶质细胞之间,胶质-神经元之间有着密切联系,形成一个错综复杂的机制网,并且仍有更多的未知领域有待研究[1]。DAI后胶质反应示意图(图1)。
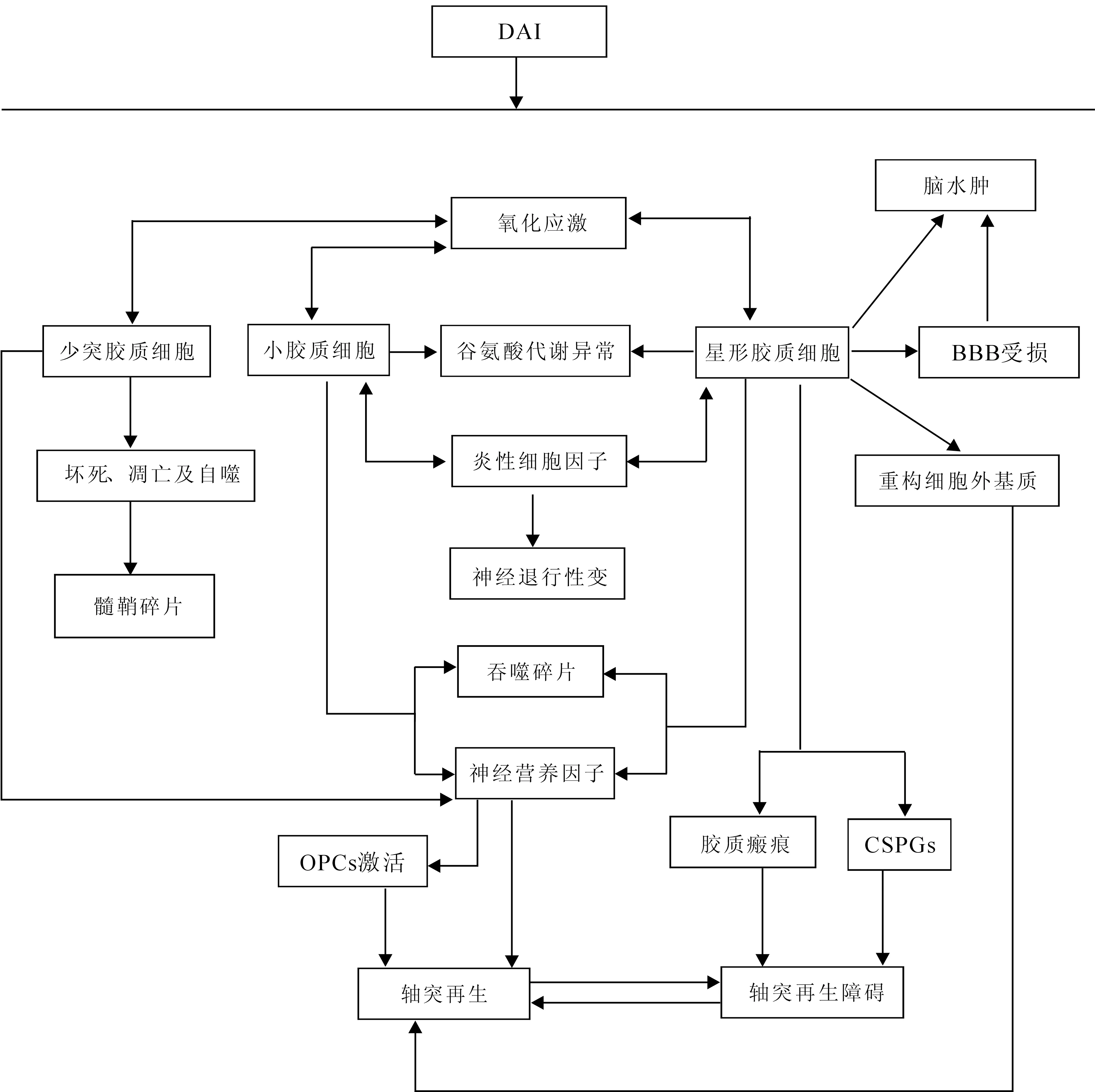
图1 DAI后胶质反应机制示意图
Fig.1 The pathophysiological mechanisms of glial reaction after DAI
2 DAI相关生化标记物的研究进展
目前,临床主要通过外伤史、症状和影像学表现诊断DAI。DAI患者伤后多即刻、长时间、深度意识障碍,无明显特异性临床特征,并且CT对非出血性病灶和针尖样大小的出血点很难识别,尽管磁共振频谱(MRS)、磁化传递成像(MTI)、磁共振弥散成像(DWI)等相关检查可以提高DAI的诊断准确率,但对于微小病灶和轻型DAI,仍存在较高的假阴性结果,且磁共振检查所需时间长,患者难以配合完成检查。此外,常规的染色方法在创伤后的12~15 h内难以发现典型的病理改变,难以诊断伤后短期存活的DAI患者。随着对DAI病理生理研究的深入,一系列来源于神经元或胶质细胞的特异性蛋白在DAI发展过程的时相规律逐渐被发现。这些蛋白在脑组织、血液和CSF中的变化反映了DAI后神经损伤和BBB破坏的程度,一定程度上可被视为诊断DAI及判断病情发展的生化指标,其中来源于轴索的蛋白,更是可直接反映轴索断裂和损伤程度。为了更早期、更准确地诊断DAI,弥补影像学及临床特征诊断DAI的不足,判断其预后,探究血浆或CSF中的DAI特异性生化指标及其诊断阈值显得尤为重要[23]。
2.1 β淀粉样前体蛋白及β淀粉样蛋白 β淀粉样前体蛋白(beta-amyloid precusor protein, β-APP)是神经元内一种具有受体样结构的跨膜糖蛋白,胞质内合成后通过轴浆运输至突触,正常情况下免疫组化技术不能检出。轴索损伤后数小时,神经元并不立即死亡,胞内细胞器仍发挥功能,β-APP mRNA上调,β-APP生成增加,加之轴浆运输受阻,大量β-APP快速聚集在轴索球,组成轴索球的外缘。因此,β-APP的免疫组化技术可早期诊断DAI[24]。但是,β-APP免疫组化染色只染出30%~50%的损伤轴索,容易让人低估实际轴索损伤范围。进一步研究发现,β-APP四种亚型之一β-APP695在正常状态下的神经元轴索中表达水平较低,神经元受损后β-APP695作为一种快速反应性蛋白表达水平明显升高,同时因轴索损伤引起轴索的轴浆运输障碍,使得β-APP695在损伤部位聚集,其也可作为诊断DAI的指标之一[25]。此外,在施旺细胞、血小板内也有β-APP存在,在缺氧、低血糖等导致轴索代谢障碍的疾病中,β-APP的表达也呈阳性,因此依靠β-APP诊断DAI,必须详细询问病史。CSF中APP的存在可能是预测DAI的指标。但是,此方向的报道不多,仍需深入研究。因此,β-APP可能不是DAI特异性标记物,其作为广泛性脑损伤的标记物可能更合适。尽管有诸多弊端,临床可操作性差,但β-APP染色仍是目前实验室及法医鉴定DAI的金标准。
目前,对β-APP衍生物的研究也取得了一定进展。β淀粉样蛋白(amyloid β, Aβ)是由β-APP经α、γ分泌酶在不同的位点剪切产生的,大量的β-APP被水解后产生Aβ聚集在核周体,Aβ的免疫染色也可以染上β-APP,伤后1 d在大脑皮层和海马可以检出Aβ,3 d后明显,6~14 d更高[26]。DAI后CSF中Aβ总量明显升高,伤后5~6 d,比正常仍高出11倍,2周后逐渐降至正常。CSF中存在Aβ(1~42)及两种前体形式,α-sAPP和β-sAPP。Aβ(1~42)在伤后5~6 d达高峰,α-sAPP在伤后7~11 d升至最高,β-sAPP也有明显的升高[27]。血浆中Aβ的量比CSF中低100倍,当CSF中Aβ变化明显时,血浆中的Aβ并没有明显变化。因此,血浆中的Aβ可能不能反映CNS中Aβ的代谢[28]。
2.2 FE65 FE65是诱导β-APP绑定在胞内域的适配器蛋白,调控β-APP易位至神经胞膜上。DAI后FE65可以诱导β-APP的聚集。FE65是高度保守的蛋白。因此,动物实验中的结果也极有可能适用于人。大鼠FE65 mRNA表达早在伤后30 min显著上调,1 h达到高峰,12 h后逐渐减少至正常,24 h时再次升高,伤后48 h再下降[29]。这种双峰现象与原发性和继发性轴索断裂的时程一致。尽管目前对FE65在轴索损伤中的变化趋势仍缺少确切的数据,但FE65及其mRNA仍可能是高度敏感、特异、并能早期诊断DAI的生化指标。
2.3 髓鞘碱性蛋白 髓鞘碱性蛋白(myelin basic protein, MBP)与髓鞘脂质紧密结合,维持CNS髓鞘结构和功能稳定,中枢性MBP由少突胶质细胞合成和分泌。DAI早期,轴索肿胀增粗,随后出现轴索断裂及髓鞘崩解,MBP的升高与轴索损害的逐渐加重及髓鞘的破坏程度基本呈平行关系。MBP被钙蛋白酶、基质金属蛋白酶、溶酶体酶等降解,伤后2 h MBP N-端降解产物开始出现,1 d后达峰,随着时间延长,坏死组织被小胶质细胞及巨噬细胞清除,7 d后恢复正常。有研究认为,脑组织中MBP降解产物作为诊断DAI的指标,具有87%的敏感性及100%的特异性[30]。DAI后CSF中MBP水平也明显升高,且无BBB的干扰,可以更加准确地反映神经细胞的损害和髓鞘损伤的程度。DAI后脑组织缺血、缺氧、坏死等继发性脑损害引起脑白质髓鞘崩解破坏加重、BBB通透性增高或破坏加重而致MBP大量进入外周血循环,血清MBP的上升可反映髓鞘等神经结构和BBB同时受损的情况,因此判断损伤和评估预后时血清MBP更具有实用性和全面性[31]。相对于通过腰穿获取CSF,检测血清中MBP更方便、安全,但血清不能及时反映病情变化,因此同步检测DAI患者血和CSF中MBP质量浓度变化,既可初步判断DAI的程度,又能间接了解BBB的受损程度,客观评估DAI的演变过程及预后,对临床用药选择也有一定的指导意义。尽管MBP被视为脑外伤的生化指标已经有30年,但是检验的敏感性限制了临床应用。CSF中检测MBP的敏感性比脑组织中要低。因此,需要研究人员开发出更加灵敏的检测手段,来量化DAI后髓鞘的损伤程度。
2.4 神经元特异性烯醇化酶 神经元特异性烯醇化酶(neuron-specific enolase, NSE)是神经元内参与糖酵解途径的烯醇化酶,主要存在于神经元及神经来源的细胞中,20世纪90年代起NSE蛋白被认为对CNS损害具有高度特异性,从而受到广泛重视。正常情况下体液中的NSE水平极低,当神经元损伤时,细胞膜完整性被破坏,由于NSE不与细胞内肌动蛋白结合,易从细胞内释放出来,通过细胞间隙进入CSF,或通过BBB进入外周血。DAI后24 h内NSE水平开始增高,2~3 d达高峰,7~14 d逐渐下降。其机制可能为轴索受损后,NSE通过受损的BBB释放入CSF及血液,随后患者开始进入恢复期,损伤逐渐趋于稳定或好转,NSE来源减少,且被持续清除,NSE开始逐渐下降。血清NSE水平在一定程度上能够间接反映CSF中NSE的变化,但由于神经元释放出的NSE被局限或被消耗,CSF中NSE较血清NSE更敏感。DAI严重程度主要与轴索损坏的程度及范围有关,破坏越严重、范围越大,释放入细胞外液中的NSE越多。因此,NSE水平与DAI的关系更紧密[32]。DAI后血清NSE浓度越高,升高持续时间越长,DAI患者病情更严重,预后不良的可能性更大。
2.5 S100B S100B蛋白是一种由脑内活化的胶质细胞分泌的酸性蛋白,在灰质中主要存在于AST,在白质则主要在少突胶质细胞中。高浓度的S100B蛋白有神经毒性作用,参与神经退化机制,细胞外高浓度的S100B蛋白可刺激致炎性细胞因子表达和细胞凋亡,并能通过一氧化氮依赖途径诱导神经元死亡。DAI患者GOS和MMSE评分与血浆S100B含量呈负相关,这提示患者颅脑损伤越严重,血浆S100B蛋白含量越高,预后越差[33-34]。在治疗期间如果S100B蛋白水平再次升高或持续在高水平,可提示继发性损伤严重,需要探究继发性脑损伤的病因并加大治疗力度,而治疗后S100B蛋白水平能迅速降低或持续维持在低水平,则提示治疗效果可靠。因此,对颅脑损伤患者治疗期间动态监测S100B蛋白水平,对于评定治疗效果和判断继发性颅脑损伤程度也具有十分重要的意义[35]。
S100B蛋白在血浆和CSF中的浓度变化也可以反映CNS损害的程度,且方便快捷,易于推广,已成为判断和评估颅脑损伤预后的特异性指标[36]。作为一种无创性的检测手段,血清S100B蛋白检测简便价廉,即使在基层医院也很容易推广普及,正日益受到临床医务工作者的重视,它作为重型颅脑损伤的监测评价指标具有重大意义。但是,除CNS外也有不少细胞可合成S100B,多系统复合性损伤患者体内即使没有颅脑损伤,其他部位的创伤,如脂肪、肌肉、骨骼等组织损伤早期也可检测到血清S100B蛋白水平的升高,但其峰值是出现在伤后1 h,且在半衰期过后则基本清除,对于急性期后血清S100B浓度几乎没有影响[37]。由于DAI患者多并发CNS外复合性损伤,所以在入院初期的S100B浓度值对病情的预期价值不高。因此,用S100B 蛋白水平来评价DAI预后的时候要考虑严重的多发伤患者的特殊情况,否则可能会造成对预后判断的不准确。
2.6 神经微丝 神经微丝(neurofilament, NF)主要位于轴突,是细胞骨架的组成部分,参与轴突运输。NF多聚合物由轻链(NF-L)、中链(NF-M)、重链(NF-H)多肽组成。正常轴索内NF侧臂是由磷酸化的NF-M和NF-H的分子末端形成的,并处于磷酸酶和蛋白激酶相互作用的动态平衡中。轴索损伤后,钙调磷酸酶调控NF去磷酸化及水解,NF侧臂被清除,三维空间发生改变。轻中度DAI轴索局部NF排列紊乱,中重度NF有致密区出现,相邻NF间空间缩小,轴浆内NF密度增加,导致磷酸化NF的稳定性降低,轴突直接减小,微管数量减少,最终导致磷酸化NF的水解,神经微丝塌陷[38]。
DAI后6 h,NF-L染色可见轴索肿胀;24 h可见轴索球数量明显增加,并且NF-L不能使胞体及树突着色[39]。因此,在NF的3种亚基中,NF-L可能是特异性和敏感性最高的轴索损伤标志物,并且NF-L在CSF中也能检测出。磷酸化的NF-H在DAI后1 d 开始减少,血清中磷酸化的NF-H碎片在伤后6 h即明显升高,12 h及48 h分别达高峰,7 d后降至正常[38]。因为继发性轴索损伤比原发性更严重,所以第2峰比第1峰更高[40]。由于血样的采集比CSF更容易,所以NF-H也是特异的DAI诊断标志物[41]。此外,亦有研究证实DAI后24 h轴突内只有少量NF-M染色,但是3 d后明显增多,关于NF-M作为DAI标记物仍需进一步研究。
2.7 微管相关蛋白tau 微管相关蛋白tau (microtubule-associated tau, MAP-tau)是一种对微管组装和功能稳定起重要作用的细胞骨架蛋白,生理状态下仅在轴索内运输。病理情况下,MAP-tau的异常磷酸化可以抑制微管组装,促进微管解聚,导致轴索的破坏及断裂。DAI后钙蛋白酶可以解聚核周和轴突的微管,MAP-tau被calpain-1及caspase-3裂解为多肽分子,即C-tau(cleaved-tau),释放至CSF和血清,伤后1 h,CSF中的C-tau可升高500~1 000倍,24 h可升高至40 000倍[42-43]。BBB的破坏导致大量的C-tau入血,由于受清除率和半衰期的影响,血浆中C-tau水平仅占CSF的十分之一,CSF与血清C-tau的差值间接反映BBB的损伤。CT上无明显征象时,DAI患者CSF中C-tau水平仍明显升高,且与临床症状改善呈负相关,表明CSF中C-tau可用于量化轴索损伤和评估临床治疗效果。预后良好(GOS评分4~5)的患者体内C-tau水平是预后差(GOS评分1~2)的十分之一,CSF中C-tau水平高于2 126 pg/mL对于预测DAI患者1年内病死率的灵敏度为100%,特异度为81.5%。因此,C-tau对于判断DAI患者预后有较高的敏感性和特异性[44]。此外,血清C-tau水平评估预后的效果并不理想,这可能是因为血清C-tau水平可能只反映急性轴索损伤[45]。C-tau是诊断DAI理想的生化标记物,分子质量小,可溶度高,其有高度的组织特异性。
2.8 血影蛋白α-Ⅱ 血影蛋白(spectrin)是一类肌动蛋白结合蛋白,神经系统内存在spectrin α-Ⅱ亚基,表达于神经元的胞体、轴突及树突中,与神经丝蛋白及MAP等共同构成神经元细胞骨架,稳定髓鞘节点,维持神经元的形态及胞膜完整性。DAI后spectrin α-Ⅱ被钙蛋白酶 (calpain-1和calpain-2)和caspase-3降解,产生血影蛋白降解产物(spectrin breakdown products, SBDPs)。分子质量280 ku的spectrin α-Ⅱ既可被降解为150 ku/145 ku或120 ku/150 ku的SBDPs。DAI后6 h在大脑皮层可检测出SBDP-145,皮髓交界处更明显,24 h SBDP水平达高峰,3 d后降低[46]。胼胝体区SBDP-145在伤后1 d升高4.2倍,3 d后达高峰,升至正常5.6倍,7 d后降至正常;SBDP-150伤后1~3 d轻度升高,但是SBDP-120没有明显的变化[47]。皮层内SBDP类型及变化趋势与胼胝体一致,表明calpain诱导的坏死是DAI后的病理生理反应[48]。DAI后CSF中SBDP-150及SBDP-145 6 h达高峰,1~3 d仍比较高。SBDP-120也在伤后6 h达高峰,伤后5 d仍较正常高。CSF中与脑组织中的SBDP升高不同步,因为蛛网膜下腔中受损的细胞可以直接释放蛋白质进入CSF,而脑实质中的细胞释放的蛋白必须经细胞间液才能转运至CSF,CSF中SBDP-145及SBDP-150与损伤的程度、病灶大小呈正相关;CSF中SBDP的类型和水平可量化轴索损伤程度、判断功能缺陷类型(局灶、弥漫)及潜在的病理机制(坏死、凋亡)[49-50]。
2.9 其他 DAI后,损伤部位环氧化酶-2表达明显增加,使来源于受损细胞膜的花生四烯酸转化为前列腺素类物质,引起活性氧的大量产生,破坏脑白质BBB,加剧脑水肿。水通透相关的AQP4参与DAI后脑水肿的形成,其表达与DAI损伤及脑水肿的变化基本一致,DAI后3 h表达增加,3 d达高峰,至10 d仍高于正常。胶质细胞释放的炎性反应因子IL-1β、IL-6、TNF等,在DAI后的级联炎性反应中起重要作用,对于判断DAI后继发损伤和炎症反应有指示作用。碱性成纤维细胞生长因子mRNA表达在24 h时增强,3 d时达高峰,并持续到7 d,拮抗损伤因子所造成的损伤,并促进组织修复。TGF-β是调节细胞生长、分化的细胞因子,在创伤愈合、免疫抑制等方面起重要作用。尽管这些蛋白不能直接反映轴索断裂程度,但这些蛋白参与DAI后脑水肿、组织碎裂、炎性反应或神经营养、修复等过程,它们与DAI的发展有着密切相关性,可作为判断DAI进展的重要辅助指标[51-52]。
3 存在的主要问题与展望
尽管目前已揭示了部分胶质细胞活化后形态及功能的改变,但无论是基础研究还是临床研究,对活化的胶质细胞如何修复或加重DAI后轴索变性的机制仍缺乏详细的了解,因此胶质细胞活化与轴索损伤之间的关系仍需进一步研究;并且长期以来对反应性胶质化在DAI中的作用也存在争论,特别是轴索损伤后小胶质/星形胶质细胞是导致神经退行性变,还是促进神经元再生尚无定论,需要更多的研究。以胶质-轴突的交互作用作为治疗靶点,重塑微观结构、促进神经修复、改善认知功能也值得进一步研究。已证实thymosinβ4、促红细胞生成素、骨髓基质细胞等均可促进OPCs的增加,诱导少突胶质细胞重生和轴突重建。尽管这些治疗手段可以改善动物的神经行为,但其对活化胶质细胞的作用靶点仍不明确,这些治疗方法如何促使胶质细胞发挥抗炎、髓鞘再生和神经修复作用的机制仍需深入探索。此外,如何加强这些治疗方案的转化医学应用,使其转化为改善人类DAI预后的新方法还需大量的临床试验。
既往研究DAI所用的主要是单种类型细胞体外培养模型,研究者常通过培养神经元模拟轴突损伤,研究神经元继发性损伤的机制;或培养单种或混合培养多种胶质细胞,研究胶质细胞之间的相互作用及其激活后的分子机制。虽然这类模型为研究DAI后单种细胞的分子病理变化提供了理论依据,但不能为研究DAI后胶质细胞-轴索之间相互作用的机制提供帮助。因此,建立神经元-胶质细胞混合培养的细胞模型,并加强器官型脑切片的应用是未来阐明胶质细胞如何促进轴索变性和修复机制的重要手段。
目前,多认为激活的胶质细胞通过直接接触和释放细胞因子间接参与DAI后神经轴索损伤“二次打击”的病理生理过程。近年来细胞间RNA转运的研究逐渐受到重视,除了神经胞体、轴索周围的胶质细胞也可能是轴索中RNA的来源,研究已证实施旺细胞-轴突之间存在核糖体转运;但其他胶质细胞-轴索间的mRNA转运仍有争议。深入研究胶质细胞和轴突细胞间的mRNA转运,对深入研究轴突胶质一体化很有帮助。如果轴索中蛋白质的合成可以由周围胶质细胞控制,那么以胶质细胞为载体的临床干预则成为可能[53]。
尽管不同的刺激会导致不同的星形胶质细胞表型变化,但是启动这一过程的分子机制仍不明确,这也是研究反应性胶质化的主要障碍之一。因此,研究者仍需深入探索星形胶质细胞活化的机制,一方面阐明星形胶质细胞活化后介导不同功能的信号通路,为以星形胶质细胞为靶点的治疗提供方向;另一方面,研究表明,不同损伤区域的星形胶质细胞有不同的异质性,但活化的星形胶质细胞是否存在类似巨噬细胞M1和M2的亚群仍不确定,星形胶质细胞种群内是否存在异质性也是未来的研究领域[3]。DAI后小胶质细胞被认为是对CNS损伤首先做出反应的细胞,神经元、胶质细胞、血管内皮细胞、血源性细胞等释放的引发小胶质细胞激活的信息分子有待于进一步澄清。此外,介导小胶质细胞活化的分子机制及相关信号通路也需深入阐明,依此可阻断小胶质细胞的受体,选择灭活转录因子,或利用基因和蛋白技术限制某些诱导基因的转录和表达,从而抑制小胶质细胞的持续激活,也可对小胶质细胞定向导入治疗基因而参与DAI的治疗。
对DAI相关的各种生化标记物的研究仍在继续,但距离临床应用仍十分遥远。一方面各种标志物诊断DAI的阈浓度还未确定,其次是病例选择、样本大小、标本采集和测定方法等也存在一定的差异;另一方面,关于DAI后神经系统相关蛋白质的时间-浓度曲线,以及其与年龄的相关性方面均未得出一致性的结论。此外,在CSF和血液中检测单一神经系统相关蛋白质的含量变化对于诊断DAI、判断预后及评估治疗效果具有一定局限性。为了增加标志物的临床应用价值,联合检测两个或多个标志物可以提高对DAI的诊断。研究者联合检测S100B及NSE认为,S100B及NSE均升高预示着原发性脑外伤,S100B升高,NSE正常则预示着多器官功能障碍。S100B作为脑损伤的生化标记物存在争议,但是S100B有很高的阴性预测价值,外伤后S100B正常可以很精确的排除脑外伤[54]。结合现有技术,综合分析多种标志物的动态变化,把DAI后核酸、蛋白的检查结合起来,而非孤立地研究某一指标,对于判断脑损伤程度、评估预后、调整治疗方案等有着重要意义。
4 结 语
近数十年关于DAI的研究集中于神经元的病理生理改变,但越来越多的证据表明DAI后胶质细胞在脑内稳态的平衡中也发挥重要作用。因此,现在对DAI的研究重点已经从既往的原发性损伤转向了继发性轴索损伤,尤其从研究神经轴索迟发型断裂的机制逐渐转变为DAI后反应性胶质化的作用和机制。目前,研究已表明DAI后激活的胶质细胞的形态和生理功能均发生改变,其一方面通过吞噬细胞碎片、隔离损伤区域、促进BBB修复、消除水肿、释放抗炎因子及多种营养因子等过程发挥脑保护作用,促进轴索及髓鞘再生;另一方面通过形成胶质瘢痕、分泌轴突生长抑制因子、释放炎性细胞因子、诱发兴奋毒性、诱导脑水肿等作用造成神经损伤及退行性变,阻碍轴突修复;胶质细胞这种“双刃剑”作用共同参与了疾病的发展。针对DAI后胶质细胞介导的脑损伤及脑保护作用,进一步揭示各种机制发生演变的规律,阐明神经-胶质细胞间的相互关系,以及深入揭示DAI相关生化标志物的意义,有助于指导未来的研究方向,为减轻DAI后脑损伤、促进脑修复提供新的视角。这些研究将对提高DAI诊断及治疗水平具有重要意义。
[1] KOU Z, VANDEVORD PJ. Traumatic white matter injury and glial activation: From basic science to clinics[J]. Glia, 2014, 62(11):1831-1855.
[2] BROSIUS LUTZ A, BARRES BA. Contrasting the glial response to axon injury in the central and peripheral nervous systems[J]. Dev Cell, 2014, 28(1):7-17.
[3] OBERHEIM NA, GOLDMAN SA, NEDERGAARD M. Heterogeneity of astrocytic form and function[J]. Methods Mol Biol, 2012, 814:23-45.
[4] ALLEN NJ, BENNETT ML, FOO LC, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors[J]. Nature, 2012, 486(7403):410-414.
[5] SOFRONIEW MV. Molecular dissection of reactive astrogliosis and glial scar formation[J]. Trends Neurosci, 2009, 32(12):638-647.
[6] ZAMANIAN JL, XU L, FOO LC, et al. Genomic analysis of reactive astrogliosis[J]. J Neurosci, 2012, 32(18):6391-6410.
[7] VOSKUHL RR, PETERSON RS, SONG B, et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS[J]. J Neurosci, 2009, 29(37): 11511-11522.
[8] ADELSON PD, JENKINS LW, HAMILTON RL, et al. Histopathologic response of the immature rat to diffuse traumatic brain injury[J]. J Neurotrauma, 2001, 18(10):967-976.
[9] BYE N, CARRON S, HAN X, et al. Neurogenesis and glial proliferation are stimulated following diffuse traumatic brain injury in adult rats[J]. J Neurosci Res, 2011, 89(7):986-1000.
[10] PFRIEGER FW. Role of glial cells in the formation and maintenance of synapses[J]. Brain Res Rev, 2010, 63(1-2):39-46.
[11] GOLDBERG JL, BARRES BA. The relationship between neuronal survival and regeneration[J]. Annu Rev Neurosci, 2000, 23:579-612.
[12] BUFFO A, ROLANDO C, CERUTI S. Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential[J]. Biochem Pharmacol, 2010, 79(2):77-89.
[13] EKMARK-LEWEN S, FLYGT J, KIWANUKA O, et al. Traumatic axonal injury in the mouse is accompanied by a dynamic inflammatory response, astroglial reactivity and complex behavioral changes[J]. J Neuroinflammation, 2013, 10:(4)44-63.
[14] WANG G, ZHANG J, HU X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury[J]. J Cereb Blood Flow Metab, 2013, 33(12):1864-1874.
[15] YU I, INAJI M, MAEDA J, et al. Glial cell-mediated deterioration and repair of the nervous system after traumatic brain injury in a rat model as assessed by positron emission tomography[J]. J Neurotrauma, 2010, 27(8):1463-1475.
[16] JIA X, CONG B, WANG S, et al. Secondary damage caused by CD11b+ microglia following diffuse axonal injury in rats[J]. J Trauma Acute Care Surg, 2012, 73(5):1168-1174.
[17] CAHOY JD, EMERY B, KAUSHAL A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function[J]. J Neurosci, 2008, 28(1):264-278.
[18] SULLIVAN GM, MIERZWA AJ, KIJPAISALRATANA N, et al. Oligodendrocyte lineage and subventricular zone response to traumatic axonal injury in the corpus callosum[J]. J Neuropathol Exp Neurol, 2013, 72(12):1106-1125.
[19] ALMAD A, SAHINKAYA FR, MCTIGUE DM. Oligodendrocyte fate after spinal cord injury[J]. Neurotherapeutics, 2011, 8(2):262-273.
[20] MAYER CL, HUBER BR, PESKIND E. Traumatic brain injury, neuroinflammation, and post-traumatic headaches[J]. Headache, 2013, 53(9):1523-1530.
[21] RANSOHOFF RM, BROWN MA. Innate immunity in the central nervous system[J]. J Clin Invest, 2012, 122(4):1164-1171.
[22] NOBUTA H, GHIANI CA, PAEZ PM, et al. STAT3-mediated astrogliosis protects myelin development in neonatal brain injury[J]. Ann Neurol, 2012, 72(5):750-765.
[23] KORFIAS S, PAPADIMITRIOU A, STRANJALIS G, et al. Serum biochemical markers of brain injury[J]. Mini Rev Med Chem, 2009, 9(2):227-234.
[24] HOSHINO S, KOBAYASHI S, FURUKAWA T, et al. Multiple immunostaining methods to detect traumatic axonal injury in the rat fluid-percussion brain injury model[J]. Neurol Med Chir (Tokyo), 2003, 43(4):165-174.
[25] MARKLUND N, FARROKHNIA N, HANELL A, et al. Monitoring of beta-amyloid dynamics after human traumatic brain injury[J]. J Neurotrauma, 2014, 31(1):42-55.
[26] SMITH DH, CHEN XH, IWATA A, et al. Amyloid beta accumulation in axons after traumatic brain injury in humans[J]. J Neurosurg, 2003, 98(5):1072-1077.
[27] OLSSON A, CSAJBOK L, OST M, et al. Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury[J]. J Neurol, 2004, 251(7):870-876.
[28] TSITSOPOULOS PP, MARKLUND N. Amyloid-beta peptides and tau protein as biomarkers in cerebrospinal and interstitial fluid following traumatic brain injury: A review of experimental and clinical studies[J]. Front Neurol, 2013, 4(6):79-96.
[29] IINO M, NAKATOME M, OGURA Y, et al. Real-time PCR quantitation of FE65 a beta-amyloid precursor protein-binding protein after traumatic brain injury in rats[J]. Int J Legal Med, 2003, 117(3):153-159.
[30] OTTENS AK, GOLDEN EC, BUSTAMANTE L, et al. Proteolysis of multiple myelin basic protein isoforms after neurotrauma: characterization by mass spectrometry[J]. J Neurochem, 2008, 104(5):1404-1414.
[31] YANG XF, WANG H, WEN L. From myelin debris to inflammatory responses: a vicious circle in diffuse axonal injury[J]. Med Hypotheses, 2011, 77(1):60-62.
[32] BERGER RP, PIERCE MC, WISNIEWSKI SR, et al. Neuron-specific enolase and S100B in cerebrospinal fluid after severe traumatic brain injury in infants and children[J]. Pediatrics, 2002, 109(2):E31-37.
[33] CASTELLANI C, BIMBASHI P, RUTTENSTOCK E, et al. Neuroprotein s-100B — a useful parameter in paediatric patients with mild traumatic brain injury?[J]. Acta Paediatr, 2009, 98(10):1607-1612.
[34] OLIVECRONA M, RODLING-WAHLSTROM M, NAREDI S, et al. S-100B and neuron specific enolase are poor outcome predictors in severe traumatic brain injury treated by an intracranial pressure targeted therapy[J]. J Neurol Neurosurg Psychiatry, 2009, 80(11):1241-1247.
[35] HALLEN M, KARLSSON M, CARLHED R, et al. S-100B in serum and urine after traumatic head injury in children[J]. J Trauma, 2010, 69(2):284-289.
[36] BOHMER AE, OSES JP, SCHMIDT AP, et al. Neuron-specific enolase, S100B, and glial fibrillary acidic protein levels as outcome predictors in patients with severe traumatic brain injury[J]. Neurosurgery, 2011, 68(6):1624-1630.
[37] DEFAZIO MV, RAMMO RA, ROBLES JR, et al. The potential utility of blood-derived biochemical markers as indicators of early clinical trends following severe traumatic brain injury[J]. World Neurosurg, 2014, 81(1):151-158.
[38] ANDERSON KJ, SCHEFF SW, MILLER KM, et al. The phosphorylated axonal form of the neurofilament subunit NF-H (pNF-H) as a blood biomarker of traumatic brain injury[J]. J Neurotrauma, 2008, 25(9):1079-1085.
[39] VAN GEEL WJ, ROSENGREN LE, VERBEEK MM. An enzyme immunoassay to quantify neurofilament light chain in cerebrospinal fluid[J]. J Immunol Methods, 2005, 296(1-2):179-185.
[40] VAJTR D, BENADA O, LINZER P, et al. Immunohistochemistry and serum values of S-100B, glial fibrillary acidic protein, and hyperphosphorylated neurofilaments in brain injuries[J]. Soud Lek, 2012, 57(1):7-12.
[41] PARK E, LIU E, SHEK M, et al. Heavy neurofilament accumulation and alpha-spectrin degradation accompany cerebellar white matter functional deficits following forebrain fluid percussion injury[J]. Exp Neurol, 2007, 204(1):49-57.
[42] CHUNG CW, SONG YH, KIM IK, et al. Proapoptotic effects of tau cleavage product generated by caspase-3[J]. Neurobiol Dis, 2001, 8(1):162-172.
[43] ZEMLAN FP, ROSENBERG WS, LUEBBE PA, et al. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins[J]. J Neurochem, 1999, 72(2):741-750.
[44] BAZARIAN JJ, ZEMLAN FP, MOOKERJEE S, et al. Serum S-100B and cleaved-tau are poor predictors of long-term outcome after mild traumatic brain injury[J]. Brain Inj, 2006, 20(7):759-765.
[45] SHAW GJ, JAUCH EC, ZEMLAN FP. Serum cleaved tau protein levels and clinical outcome in adult patients with closed head injury[J]. Ann Emerg Med, 2002, 39(3):254-257.
[46] KOBEISSY FH, OTTENS AK, ZHANG Z, et al. Novel differential neuroproteomics analysis of traumatic brain injury in rats[J]. Mol Cell Proteomics, 2006, 5(10):1887-1898.
[47] CARDALI S, MAUGERI R. Detection of alphaII-spectrin and breakdown products in humans after severe traumatic brain injury[J]. J Neurosurg Sci, 2006, 50(2):25-31.
[48] BROPHY GM, PINEDA JA, PAPA L, et al. alphaII-Spectrin breakdown product cerebrospinal fluid exposure metrics suggest differences in cellular injury mechanisms after severe traumatic brain injury[J]. J Neurotrauma, 2009, 26(4):471-479.
[49] MCGINN MJ, KELLEY BJ, AKINYI L, et al. Biochemical, structural, and biomarker evidence for calpain-mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion[J]. J Neuropathol Exp Neurol, 2009, 68(3):241-249.
[50] PINEDA JA, LEWIS SB, VALADKA AB, et al. Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury[J]. J Neurotrauma, 2007, 24(2):354-366.
[51] TATE CM, WANG KK, EONTA S, et al. Serum brain biomarker level, neurocognitive performance, and self-reported symptom changes in soldiers repeatedly exposed to low-level blast: a breacher pilot study[J]. J Neurotrauma, 2013, 30(19):1620-1630.
[52] HONDA M, TSURUTA R, KANEKO T, et al. Serum glial fibrillary acidic protein is a highly specific biomarker for traumatic brain injury in humans compared with S-100B and neuron-specific enolase[J]. J Trauma, 2010, 69(1):104-109.
[53] SOTELO JR, CANCLINI L, KUN A, et al. Glia to axon RNA transfer[J]. Dev Neurobiol, 2014, 74(3):292-302.
[54] NAEIMI ZS, WEINHOFER A, SARAHRUDI K, et al. Predictive value of S-100B protein and neuron specific-enolase as markers of traumatic brain damage in clinical use[J]. Brain Inj, 2006, 20(5):463-468.
(编辑 韩维栋)
Advances in research on glial response and the biomarkers associated with diffuse axonal injury
SONG Jin-ning, ZHAO Yong-lin
(Department of Neurosurgery, the First Affiliated Hospital, Medical School of Xi’an Jiaotong University, Xi’an 710061, China)
Glial response has been proved to be closely related to secondary injury, axonal regeneration and prognosis of patients, and several specific biochemical markers reveal significant changes after diffuse axonal injury (DAI). The existing studies show that glial cells are involved in secondary axonal injury by glial scar formation, neuro-inflammation and loss of myelin; and also in promoting axonal repair and regeneration by releasing neurotrophic factors, clearing cell debris and preventing the spread of lesions after DAI. The interactions of glial cells with glial cells and glial cells with neurons ultimately result in the death of neurons and secondary axonal injury. Specific biochemical markers, such as β-amyloid precursor protein, neurofilament and C-tau, can indicate the occurrence and development of DAI earlier and more accurately, and will make up for the defects of diagnostic imaging and clinical features of DAI. These studies not only provide therapeutic target for nerve protection and axonal regeneration after DAI, but also benefit further studying and looking for new DAI-related specific biochemical markers. Further exploring the molecular mechanisms of activation of glial cells, glial-glial interaction and glial-neuron interaction, revealing the evolvement of these complex pathophysiological processes and the potential function of DAI-related biochemical markers are of great significance in reducing brain injury, promoting axonal regeneration and improving the ability of diagnosis and treatment of DAI.
traumatic brain injury; diffuse axonal injury; glial response; biochemical marker
2014-07-21
2014-10-07
国家自然科学基金资助项目(No.30471774);教育部新世纪优秀人才支持计划资助项目(No.NCET-05-0831);陕西省自然科学基金资助项目(No.2003C1-16) Supported by the National Natural Science Foundation of China (No.30471774), the New-Century Excellent Talents Program of Ministry of Education (No.NCET-05-0831), and the Natural Science Foundation of Shaanxi Province (No.2003C1-16)
宋锦宁. E-mail: jinnings@126.com
R651.1+5
A
10.7652/jdyxb201501001
优先出版:http://www.cnki.net/kcms/detail/61.1399.R.20141118.1536.001.html(2014-11-18)
