高效分子排阻色谱法测定注射用头孢匹胺钠中聚合物的含量
2015-02-23郭艳娟郭福庆
郭艳娟,郭福庆
(天津市药品检验所,天津 300070)
高效分子排阻色谱法测定注射用头孢匹胺钠中聚合物的含量
郭艳娟,郭福庆
(天津市药品检验所,天津 300070)
目的:建立高效分子排阻色谱法测定注射用头孢匹胺钠中聚合物的含量。方法:采用TSK-GEL G2000SWXL色谱柱(7.8 mm×300 mm);以磷酸盐缓冲液(pH 7.0)[0.01 mol/L磷酸氢二钠-0.01 mol/L磷酸二氢钠溶液(61∶39)]-乙腈(97.5∶2.5)为流动相;检测波长231 nm;柱温30 ℃;流速0.8 ml/min;进样量10 μl。结果:头孢匹胺在1.962 5~19.624 5 μg/ml范围内与峰面积呈良好的线性关系(r=1.000 0),最低检出浓度0.08 μg/ml。结论:建立的方法简单、快速、灵敏度高,可有效控制产品的质量。
注射用头孢匹胺钠,高分子聚合物,TSK-GEL G2000SWXL凝胶色谱柱
注射用头孢匹胺钠(cefpiramide sodium)近年来广泛应用于临床,对革兰阳性菌有很强的抗菌作用,对革兰阴性菌在内的细菌亦有广谱抗菌活性。同时,对绿脓杆菌等葡萄糖非发酵革兰阴性杆菌有很强的抗菌活性,本药作用为杀菌,并对各种细菌产生的β-内酰胺酶稳定。研究证实,β-内酰胺类抗生素中的高分子聚合物是引发速发型过敏反应的过敏原[1,2],且药品中高分子聚合物的含量直接影响过敏反应的发生率。故控制产品中的高分子聚合物的含量,对减少此类抗生素过敏具有重要意义[3]。在传统的聚合物控制中,多采用葡聚糖凝胶G10色谱柱分析。有文献报道[4],注射用头孢匹胺钠也采用葡聚糖凝胶柱进行聚合物的测定,但该法专属性差,分离效率低,灵敏度低,分析时间长。近年来多采用TSK凝胶色谱柱,即以球状蛋白亲水凝胶为填充剂的一系列新型色谱柱替代进行聚合物的分析。相较G10色谱柱,TSK凝胶柱具有分析时间短,灵敏度高等特点。本文通过方法学验证建立了采用TSK凝胶色谱柱测定头孢匹胺钠高分子聚合物的HPLC法,采用此方法对3批注射头孢匹胺钠进行了检验,并与其他现有文献方法进行了比较,验证了检测方法的可行性。
1 仪器与试药
Agilent 1100液相色谱仪(美国Agilent公司),Mettler Toledo XS 205 型电子分析天平(瑞士梅特勒公司)。注射用头孢匹胺钠(韩国某厂生产,规格为1.0 g,批号4001、4003、4006)。头孢匹胺钠对照品(韩国某厂提供,批号S14-251,纯度:93.7%)、5-巯基-1-甲基四氮唑和苯甲酸钠(韩国某厂提供)、磷酸氢二钠、磷酸二氢钠均为分析纯,乙腈为色谱纯,水为纯化水。
2 方法与结果
2.1 色谱条件 TSK-GEL G2000SWXL色谱柱(7.8 mm×300 mm),流动相采用磷酸盐缓冲液(pH 7.0)[0.01 mol/L磷酸氢二钠-0.01 mol/L磷酸二氢钠溶液(61∶39)]-乙腈(97.5∶2.5),流速为0.8 ml/min,检测波长为231 nm,柱温30 ℃,进样量10 μl。照分子排阻色谱法(《中国药典》2010年版二部附录Ⅴ H)测定。
2.2 溶液的配制
2.2.1 供试品溶液 取注射用头孢匹胺钠适量,精密称定,加水溶解并定量稀释成每1 ml中约含头孢匹胺1.0 mg的溶液,作为供试品溶液。
2.2.2 对照品溶液 取头孢匹胺对照品适量,精密称定,加流动相溶解并定量稀释制成每1 ml中约含100 μg的溶液,精密量取5 ml置50 ml量瓶,用水稀释至刻度,摇匀,作为对照溶液。
2.2.3 定位溶液 取5-巯基-1-甲基四氮唑10 mg,置100 ml量瓶,加甲醇1 ml使溶解,用水稀释至刻度,摇匀,精密量取1 ml置10 ml量瓶,用水稀释至刻度,作为定位溶液。
2.3 系统适用性试验 取5-巯基-1-甲基四氮唑10 mg,置100 ml量瓶,加甲醇1 ml使溶解,用水稀释至刻度,摇匀,精密量取1 ml置10 ml量瓶,加“2.2.1”项下供试品溶液稀释至刻度,摇匀,取10 μl注入液相色谱仪,记录色谱图,5-巯基-1-甲基四氮唑峰与其前相邻杂质峰的分离度为3.3(应大于1.5),见图1。另精密量取对照溶液5 ml置200 ml量瓶,用水稀释至刻度,摇匀,作为灵敏度溶液,取灵敏度测试溶液10 μl注入液相色谱仪,主成分峰高的信噪比为15(应大于10)。
2.4 专属性试验
2.4.1 酸破坏 取“2.2.1”项下供试品溶液10 ml,加0.1 mol/L的盐酸溶液1 ml,水浴加热1 min后,放置至室温,加0.1 mol/L的氢氧化钠溶液1 ml,摇匀。
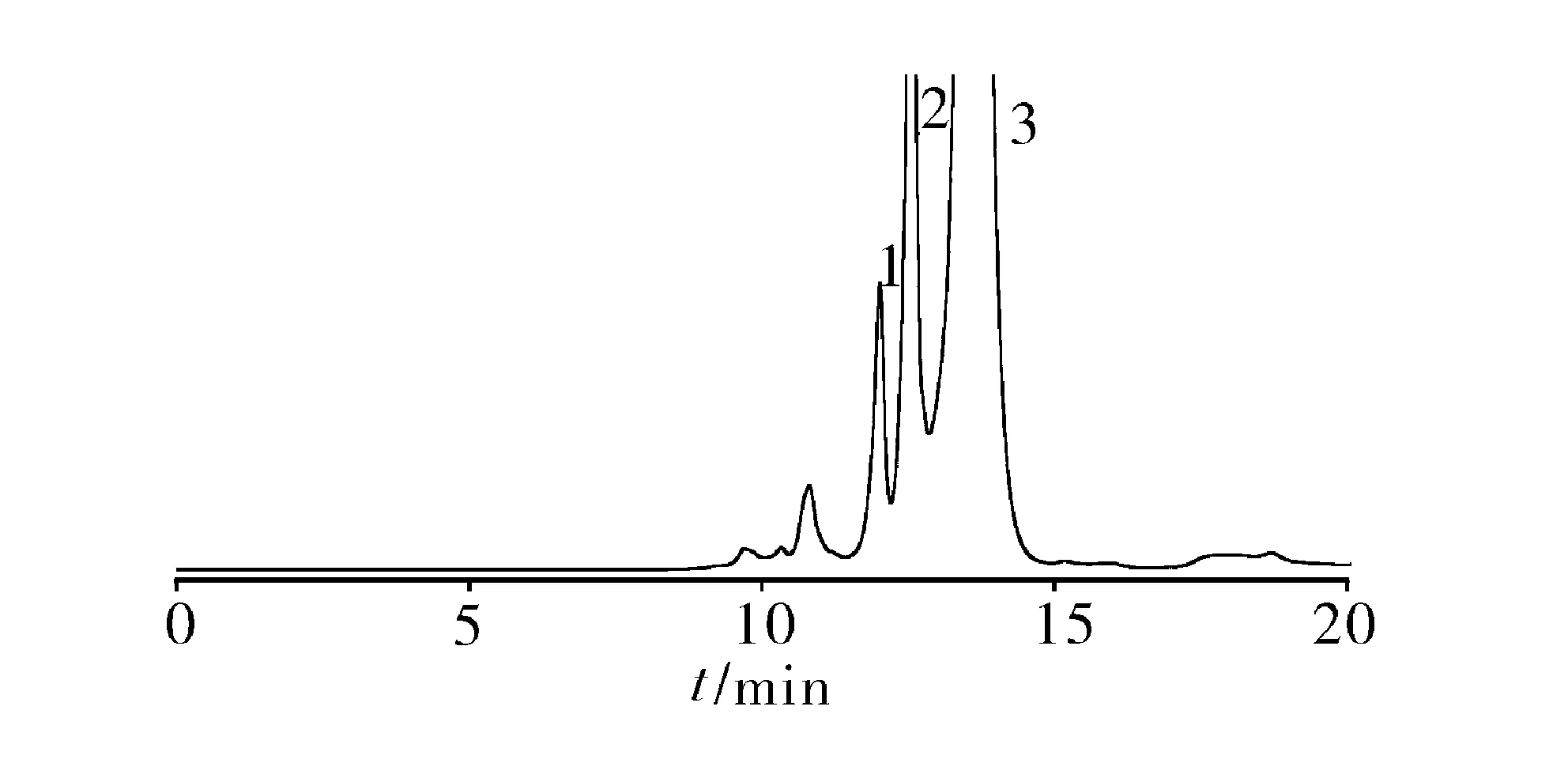
1.5-巯基-1-甲基四氮唑 2.苯甲酸钠 3.头孢匹胺钠
2.4.2 碱破坏 取“2.2.1”项下供试品溶液10 ml,加0.1 mol/L的氢氧化钠溶液1 ml,室温放置3 min后,加0.1 mol/L盐酸溶液1 ml,摇匀。
2.4.3 氧化破坏 取“2.2.1”项下供试品溶液10 ml,加1%的过氧化氢溶液1 ml,室温放置4 min。
2.4.4 加热破坏 取“2.2.1”项下供试品溶液10 ml,置100 ℃水浴加热1 min后,放置至室温。
2.4.5 光照破坏 取“2.2.1”项下供试品溶液10 ml,置254 nm紫外灯下光照30 min。
取酸、碱、氧化、加热和光照破坏后的溶液,分别进样分析,结果见图2。聚合物峰间、聚合物峰与其他杂质峰间均能良好分离,方法专属性好。
2.5 线性关系考查 取头孢匹胺钠对照品26.18 mg置250 ml量瓶,用流动相溶解并稀释至刻度,摇匀,作为对照储备液,分别取上述对照储备液1、2、4、5、6、8和10 ml置50 ml量瓶,加水稀释至刻度,摇匀,依法测定。结果表明:对照溶液浓度在1.962 5~19.624 5 μg/ml范围内,溶液浓度与测得的峰面积呈良好的线性关系,回归方程Y=31.688X+0.597 6(r=1.000 0)。
2.6 样品取样量与聚合物测定结果的相关性考查 取4006批注射用头孢匹胺钠样品约60、120和180 mg,分别置3个100 ml量瓶中,依法测定,计算保留时间小于5-巯基-1-甲基四氮唑峰的各杂质峰面积的和,结果表明,供试品取样量在62.97~184.97 mg范围内,取样量与聚合物峰面积总和呈良好线性关系,回归方程为:Y=2.794 3X+4.990 5(r=1.000 0)。
2.7 最低检出限 经试验测试最低检测浓度为0.08 μg/ml,其峰高为噪音的3倍,计算最低检测限为0.008%。
2.8 重复性试验 取4006批注射用头孢匹胺钠样品,照“2.2.1”项下方法配制6份供试品溶液,进样分析,计算保留时间小于5-巯基-1-甲基四氮唑峰的各杂质峰面积的和,结果的RSD为1.4%,表明重复性良好。
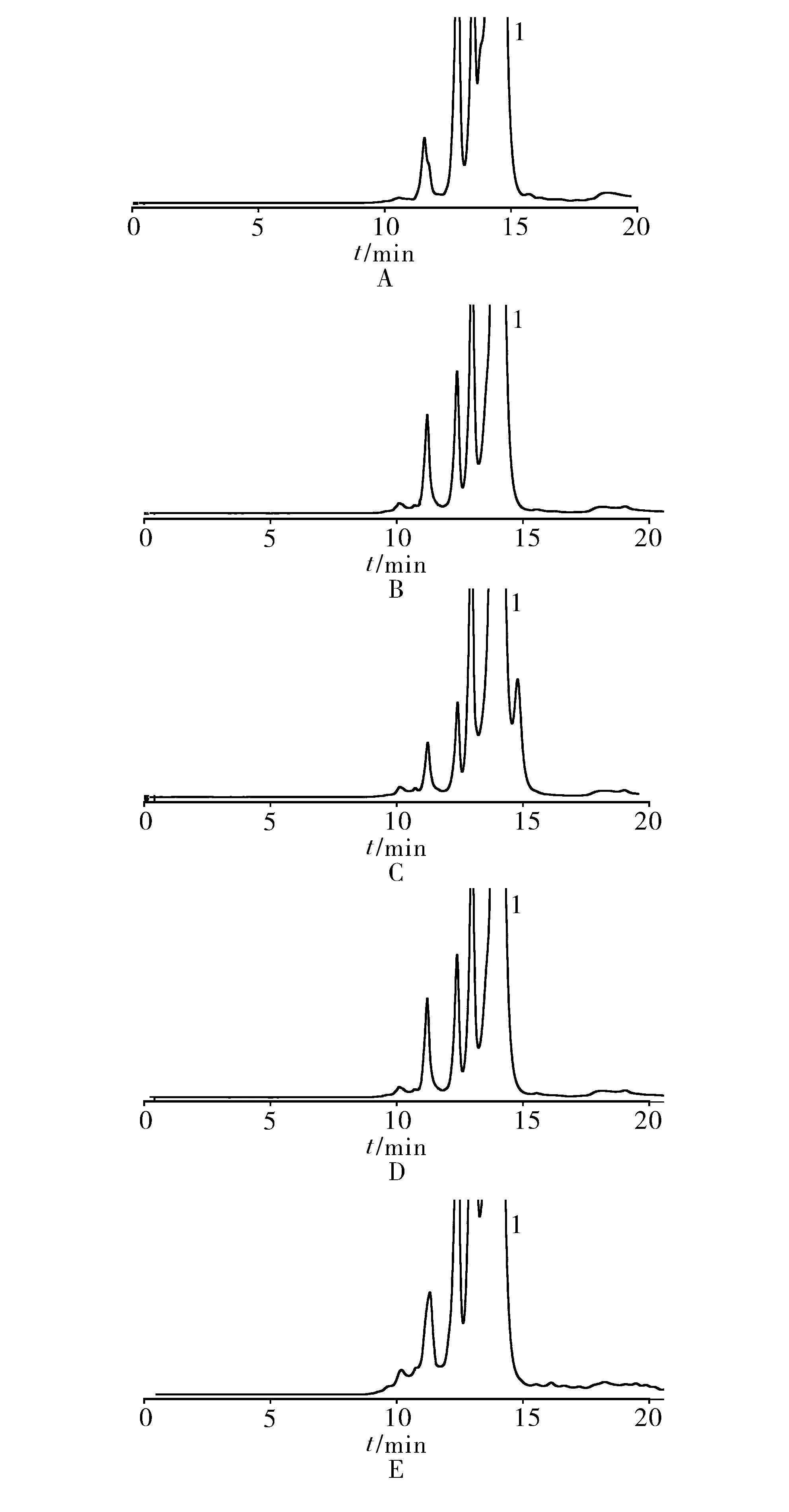
1.头孢匹胺钠
2.9 稳定性试验 取4006批样品制成供试品溶液分别于4 ℃条件下放置23 min及1、2、3、4、5、6、7和8 h后进样测试,计算保留时间小于5-巯基-1-甲基四氮唑峰的各杂质峰面积的和,结果表明,杂质总和随放置时间的延长逐渐增加,见表1,因此供试品溶液配制好后应于低温保存并尽快进样分析。
2.10 样品测定结果 取注射用头孢匹胺钠4001、4003和4006批样品,依法测定,计算保留时间小于5-巯基-1-甲基四氮唑峰的各杂质峰面积的和,总杂质结果分别为0.96%、0.94%和0.91%。

表1 稳定性试验结果
3 讨论
3.1 流动相的选择 通过以磷酸盐缓冲液(pH 7.0)[0.005 mol/L磷酸氢二钠-0.005 mol/L磷酸二氢钠溶液(61∶39)]-乙腈(95:5)、磷酸盐缓冲液(pH 7.0)[0.005 mol/L磷酸氢二钠-0.005 mol/L磷酸二氢钠溶液(61∶39)]-乙腈(97.5∶2.5)、磷酸盐缓冲液(pH 7.0)[0.01 mol/L磷酸氢二钠-0.01 mol/L磷酸二氢钠溶液(61∶39)]-乙腈(97.5∶2.5)为流动相进行比较试验,结果以磷酸盐缓冲液(pH 7.0)[0.01 mol/L磷酸氢二钠-0.01 mol/L磷酸二氢钠溶液(61∶39)]-乙腈(97.5∶2.5)为流动相时,分离效果最好,故选用磷酸盐缓冲液(pH7.0)[0.01 mol/L磷酸氢二钠-0.01 mol/L磷酸二氢钠溶液(61∶39)]-乙腈(97.5∶2.5)为流动相。
3.2 波长的选择 以231 nm波长测定结果各杂质峰的响应值均高于以254 nm波长测定,灵敏度高,故选用231 nm波长测定。
3.3 不同方法的比较 采用TSK凝胶色谱柱进行分析时,主峰前出现的杂质峰中除聚合物峰外,也可能存在其他非聚合物的杂质峰,本文通过用已知杂质对照品在相同色谱条件下进样发现,主峰前保留时间12.145 min的峰为5-巯基-1-甲基四氮唑(为头孢匹胺的主要降解杂质),保留时间12.687 min的峰为苯甲酸钠(辅料峰),故计算聚合物含量时应将这两个峰去除。否则,会造成测定结果偏高。相关文献[5]未将主峰前非聚合物的杂质峰去除,故造成测定结果远高于采用传统葡聚糖凝胶色谱柱的相关文献[4]。采用本文方法只计算保留时间小于5-巯基-1-甲基四氮唑峰的各杂质峰面积的和采用传统葡聚糖凝胶色谱柱的相关文献[4]方法测定4001批样品结果分别为0.96%和0.94%,基本一致。
综上所述,本法比其他文献方法更能准确、客观、快速、有效的测定注射用头孢匹胺钠中聚合物的含量。
1 邓增潮. 关于头孢菌素类药物用药前做皮试的探讨[J].现代医药卫生,2005,21(11):1449
2 蔡姗英, 胡昌勤. β-内酸胺类抗生素中高分子聚合物的分离分析方法研究进展[J].广东药学院学报,2002,18(2):138-142
3 袁雯玮. 高分子聚合物研究与中国药典2005年版β-内酰胺类抗生素高分子聚合物修订情况及操作要点[J].中国抗生素杂志,2005,30(12): 727-730
4 汪梅,朱美荣. 注射用头孢匹胺钠中头孢匹胺聚合物的测定[J].现代食品与药品杂志, 2007, 30(12): 727
5 徐明琴,陈林,张静霞,等. HPSEC法测定注射用头孢匹胺钠中聚合物的含量[J].国外医药抗生素分册, 2014, 35(3): 131-133
Determination of polymers in cefpiramide sodium for injection by HPSEC
Guo Yanjuan,Guo fuqing
(Tianjin institute for Drug Control,Tianjin 300070)
Objective: To establish an HPSEC method for determination of the polymer in cefpiramide sodium for injection. Method: The analysis was carried out using a TSK-GEL G2000SWXLchromatographic column (7.8 mm×300 mm) with the mobile phase containing 97.5% of acetonitrile and 2.5% of 0.01 mol/L phosphate buffer solution [containing 0.01 mol/L disodium hydrogen phosphate and 0.01 mol/L sodium dihydrogen phosphate(61∶39),pH 7.0]. The detective wavelength was set at 231 nm and the temperature of the column at 30 ℃.The flow rate was 0.8 ml/min with 10 μl of the sample to be injected into the HPLC. Results: The linear range of cefpiramide sodium was 1.9625~19.6245 μg/ml(r=1.000 0),and LOD was 0.08 μg/ml. Conclusion: The method is simple and selective for controlling the quality of the product effectively.
cefpiramide sodium for injection,polymer,TSK-GEL G2000SWXLcolumn
2014-11-18
R927.2
A
1006-5687(2015)02-0021-04
