膜超极化激活离子通道及其靶向抗癫痫药物研究进展
2015-02-01孙智明黄卓
孙智明,黄卓*
(1.北京大学药学院分子与细胞药理学系天然药物与仿生药物国家重点实验室,北京 100191;2.中国科学院上海药物研究所新药研究国家重点实验室,上海 201203)
·前沿与进展·
ADVANCES IN PHARMACEUTICAL SCIENCES
膜超极化激活离子通道及其靶向抗癫痫药物研究进展
孙智明1,2,黄卓1,2*
(1.北京大学药学院分子与细胞药理学系天然药物与仿生药物国家重点实验室,北京 100191;2.中国科学院上海药物研究所新药研究国家重点实验室,上海 201203)
癫痫是一种较为常见的神经系统疾病,主要以大量神经元同步异常放电为特征。目前普遍认为,神经元或神经网络兴奋性和抑制性电信号传输的失衡,是癫痫发病的最根本原因。现有的抗癫痫药物主要以钠离子通道、钙离子通道、钾离子通道、谷氨酸受体和γ-氨基丁酸离子通道为靶点,但接受这些药物治疗后,仍有近1/3的病人无法控制癫痫发作。因此,抗癫痫药物的研发亟需新靶点和新思路。许多研究证据表明,膜超极化激活离子通道的基因突变可以导致遗传型癫痫的发作,且在脑部损伤后,膜超极化激活离子通道会发生表达水平、通道生物物理学性质及通道亚基构成的改变,从而增加神经元和神经网络兴奋性,促使癫痫发病。故近年来,膜超极化激活离子通道及其靶向抗癫痫药物研究引起人们广泛关注。综述膜超极化激活离子通道与癫痫发病之间的关系,并探讨以膜超极化激活离子通道为靶点进行抗癫痫药物开发和治疗的可行性。
膜超极化激活离子通道;癫痫;抗癫痫药物
癫痫是一种脑功能失常综合征,主要由脑内大量神经元同步异常放电所引起,其全球发病率约为1%。在中国,癫痫是神经科发病率仅次于神经痛的第2大疾病,约影响1 500万人口。癫痫的病因复杂,目前认为离子通道基因突变、代谢紊乱、氧化应激、感染、抑制性神经元凋亡等因素均能引起癫痫的发作[1],但科学界普遍认为,神经元或神经网络兴奋性和抑制性电信号传输的失衡是导致癫痫发病的最根本原因[2]。
神经元的电信号由离子通道产生。兴奋性离子通道(如钠离子通道或钙离子通道)可让正电荷流入细胞,从而导致神经元去极化,而抑制性离子通道[如钾离子通道或γ-氨基丁酸(GABA)离子通道]则能让正电荷流出细胞或负电荷流入细胞,从而使神经元超极化。到目前为止,市场上抗癫痫药物主要以钠离子通道、钙离子通道、钾离子通道、谷氨酸受体及GABA离子通道为靶点[1]。接受这些药物治疗后,虽然大部分病人能达到控制癫痫发作的效果,但仍有近1/3的病人无法控制癫痫的发作[3],故只能通过手术治疗,切除癫痫发作的原发区域,而术后病人往往会有出现不同程度的认知功能障碍,影响生活质量。再者,这些药物副作用较大,可致病人产生眩晕、头痛、精神紧张等症状,而且通常不能有效阻止病情的发展[4],长期使用还会导致耐药性产生,形成顽固性癫痫。此外,儿童长期使用这些药物后,通常会导致智力发育迟缓、认知功能障碍等不良反应。因此,抗癫痫领域亟需发现新的靶点,为新一代抗癫痫药物的研发提供新思路。
膜超极化激活离子通道(hyperpolarization-activated cation non-selective channel, HCN通道)是电压依赖型阳离子非选择性通道,通常在细胞膜电压小于-70 mV时开放,可让钠离子、钾离子等一价阳离子(除锂离子)通过,但二价阳离子不能通过;该通道电流被称为Ih电流。目前已有共4种HCN通道的亚基被克隆,即HCN1~HCN4[5-7],每种HCN亚基由六次跨膜蛋白构成,4个亚基构成一个HCN通道,它们可形成同源或异源四聚体。在中枢神经系统中,主要表达HCN1和HCN2两种亚基,它们有着明显的区域化分布的特征,其中,HCN1大量分布于新皮层、海马体和小脑;HCN2在大部分脑区内均有表达;HCN3在脑内低表达,主要分布于下丘脑和延髓等部位;HCN4主要分布于丘脑核、基底神经节和嗅球。与HCN1相比,HCN2表现出较慢的激活与失活特性。此外,与HCN3和HCN4相比较,HCN1和HCN2对细胞内环磷腺苷(cAMP)浓度不敏感[8]。近10年来,越来越多的研究表明,脑内HCN通道表达或功能的变化可能直接导致癫痫的发生和发展[9-15]。因此,以HCN通道为靶点开发抗癫痫药物,引起广泛关注。
1 HCN通道与癫痫
癫痫可分为遗传型和继发型两种,遗传型癫痫主要由离子通道、代谢、神经传导通路等相关基因突变所导致,而继发型癫痫主要是由于脑外伤、中风、肿瘤、感染等脑部损伤因素所引起。
1.1 HCN通道与遗传型癫痫
越来越多的证据表明,HCN通道的基因突变可以诱导癫痫的发作。在对癫痫病人家庭成员进行外显子测序时发现,HCN1存在错译的点突变。且在体外研究中发现,这些错译突变会直接影响到通过HCN1的电流,而Ih电流的增加或减少均能诱导癫痫的发作[16]。此外,研究人员在遗传型癫痫病人或先天癫痫发作小鼠组织中检测到HCN2基因突变位点,这些基因位点的突变或是影响了HCN2调节的Ih电流,或是直接降低了HCN2 mRNA及其蛋白的表达水平,从而导致失神性癫痫的发生和发展[17]。
1.2 HCN通道与继发型癫痫
对于继发型癫痫来讲,脑部损伤后,病人在最初的几个月到几年内通常并无癫痫发作,此阶段被叫做癫痫的潜伏期,而在潜伏期,神经元和神经网络会发生一系列的亚细胞变化,从而导致潜伏期过后病人神经元细胞过度兴奋及异常放电,引起反复的自发性癫痫发作(见图1)。目前科学界普遍认为,在颞叶癫痫的潜伏期,神经元和神经网络兴奋性的增加是导致自发性癫痫发作的最主要原因[18]。脑损伤可引起内嗅皮层椎体细胞上HCN通道的表达发生变化,从而导致神经元兴奋性增高。持续性癫痫发作1、7和14 d后,树突和神经末梢上HCN通道的表达明显降低,而树突上HCN通道表达的降低引起输入电阻增加以及树突上兴奋性突触后电流(EPSC)的增加,神经末梢上HCN通道表达的降低则引起T型钙离子通道(T channels)功能增强,致使EPSC传输频率增加。这些变化显著增强了内嗅皮层椎体细胞兴奋性,促使自发性癫痫的发作(见图2)[13-14,19]。
目前有3种广泛用于模拟继发型癫痫发病过程的鼠科动物模型,这些动物模型采用不同的方法(如电刺激、药物诱导等)来模拟脑部损伤,并诱导持续性癫痫的发作。由此观察到,在脑损伤和持续性癫痫发作之间呈现明显的潜伏期特征[20]。
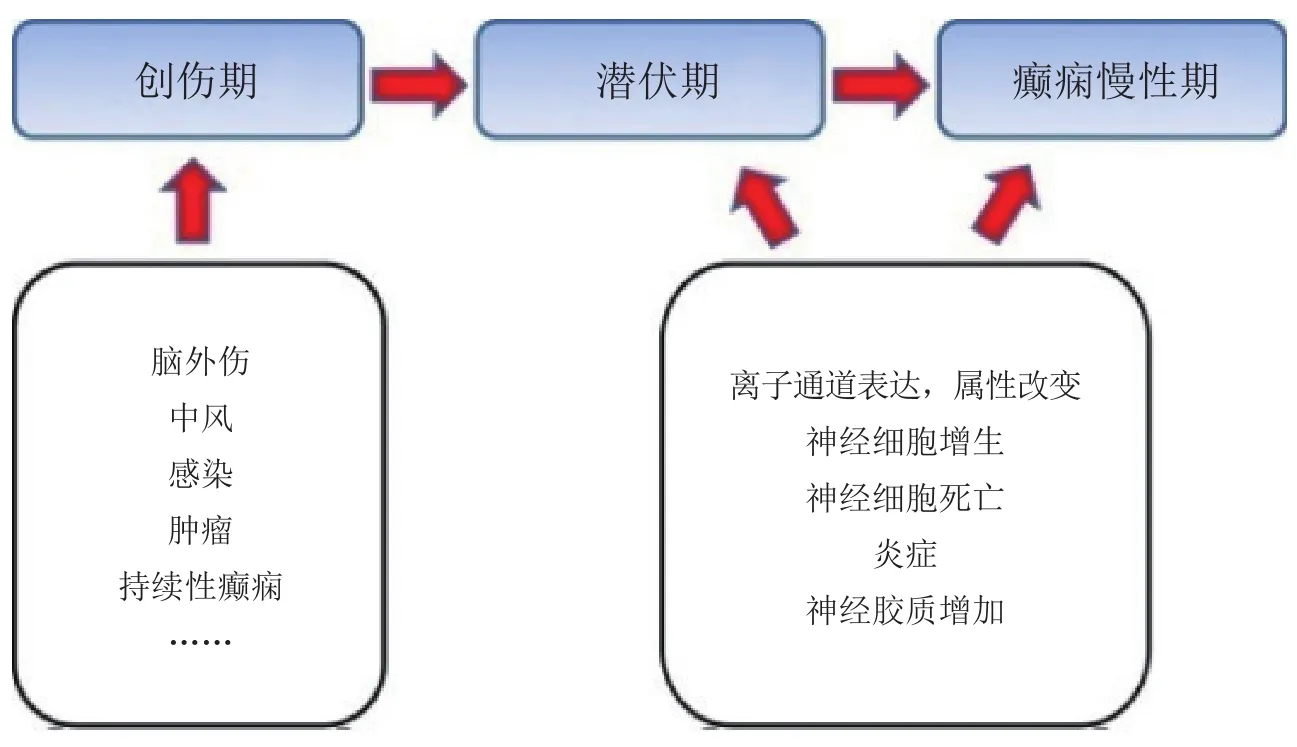
图1 继发型癫痫的发病过程Figure 1 Pathogenesis of secondary epilepsy
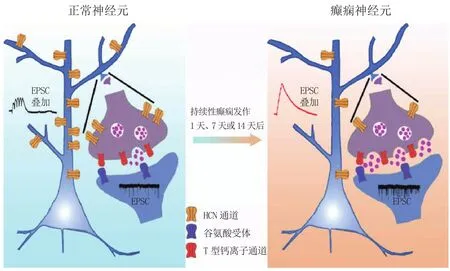
图2 HCN通道与继发型癫痫的关系Figure 2 Relationship between HCN channels and secondary epilepsy
动物模型实验显示,在癫痫潜伏期,中枢神经系统中HCN通道会发生表达水平、通道生物物理学性质及通道亚基构成的改变(见表1)。首先,脑损伤会在蛋白转录和翻译水平上影响HCN通道的表达水平,如在海马体中,持续性癫痫可减少HCN1和HCN2 mRNA及其蛋白的表达水平[21-25]。其次,一些HCN通道的附属亚基,如MiRP1、S-SCAM、Mint2、Filamin A、vitronectin等,特别是最近鉴别出的TRIP8b[tetratricopeptide repeat (TPR)-containing Rab8b],被证实可调节HCN通道的表达[13,30-36],而在癫痫潜伏期,持续性癫痫会干扰TRIP8b与HCN1的相互作用,从而降低HCN1的表达[13,37]。第三,在癫痫潜伏期,HCN1的蛋白磷酸化水平发生变化,而钙调磷酸酶(calcineurin)活性升高和p38 MAPK激酶活性降低,直接导致HCN1的电流减弱,促使癫痫发作[28,38-39]。最后,HCN通道的糖基化过程决定了HCN 通道的亚基构成,大量的HCN1糖基化致使HCN1/HCN2的表达水平增加,改变了HCN通道的性质和正常的生理功能[29,40-41]。提示,HCN通道的糖基化过程可能同样与癫痫的发生和发展有着密切关系。
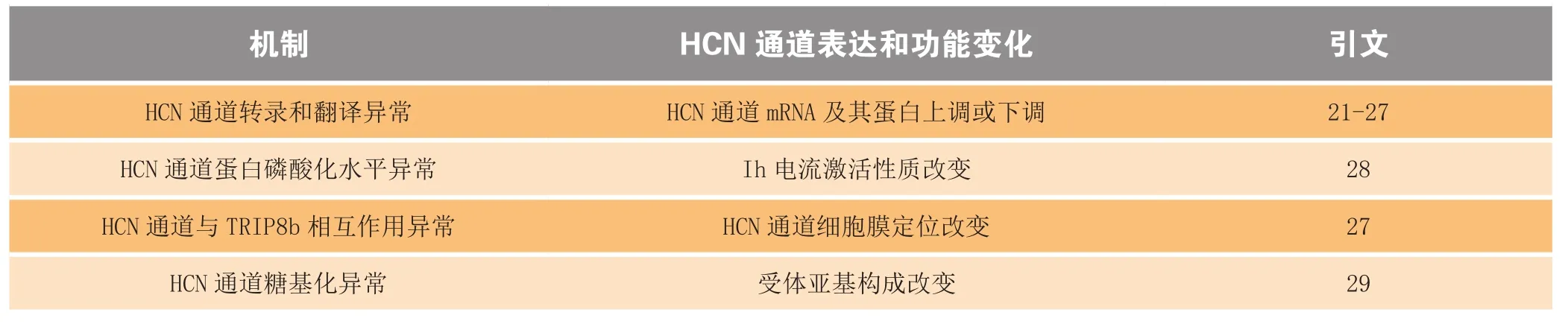
表1 脑损伤后HCN通道的表达和功能变化机制Table1 Mechanisms of changes in the expression and function of HCN channels after brain injury
那么,在癫痫潜伏期HCN通道的表达和功能变化是否是癫痫发作的决定性因素?越来越多的研究证据给出的答案是肯定的。HCN通道表达或功能的增加或降低均能引起神经元兴奋性升高。例如,在颞叶癫痫的潜伏期,树突上HCN通道表达的降低致使树突上兴奋性突触后电位(EPSP)升高,从而增加了神经元产生动作电位的机率,增强了神经元的兴奋性;此外,树突上HCN通道表达的降低还会显著增加神经元的输入电阻,使相同输入电流产生更大的突触后电位变化,也增强了神经元的兴奋性[21];但是,在大鼠热惊厥(febrile seizure)模型中,体温升高可导致海马体CA1锥体细胞上HCN通道的表达增加,并致使高频的抑制性突触后电位激活Ih电流,进而产生非正常的回弹放电(rebound firing),同样也增强了神经元的兴奋性[24]。
现有的研究结果表明HCN通道在兴奋性和抑制性神经元上均有表达,但目前的研究主要集中在兴奋性神经元上,而抑制性神经元上HCN通道的功能尚不十分清楚。在内嗅皮层,HCN1选择性定位于兴奋性末梢的突触前膜上,而特异性阻断这些突触前膜上HCN1,能明显增加兴奋性神经递质的传输频率,导致神经网络兴奋性的提升[14]。在颞叶癫痫的潜伏期,兴奋性突触前膜上HCN1的表达明显降低,增强了兴奋性神经递质的传输,影响了兴奋性和抑制性的平衡,促使癫痫发作(见图2)[13-14,19]。而抑制性神经递质不受这种调节作用影响,说明在内嗅皮层中HCN1仅存在于兴奋性神经末梢。此外,HCN2基因敲除小鼠表现出失神性癫痫的表型[42]。丘脑是失神性癫痫的主要原发区域,丘脑皮层系统(thalamocortical system, TC)神经元主要表达HCN2和HCN4,这些亚基通道具有较慢的开放和关闭的生物物理学特征及很高的cAMP敏感性,而缺少HCN2的TC神经元会将单个动作电位的放电模式转变为群发放电模式(burst firing),从而增加神经网络的同步性,促进失神性癫痫的发作[42-43]。
2 以HCN通道为靶点进行抗癫痫药物开发与治疗的可行性
鉴于HCN通道表达和功能的增强或减弱均能导致神经元兴奋性异常,从而诱导癫痫发作,因此将HCN通道作为抗癫痫靶点,以恢复HCN通道的正常生理学功能,不失为抗癫痫药物研发的方向之一。我们可以基于HCN通道表达和功能失常的机制而发掘药物靶点。例如,TRIP8b是调节HCN通道运输和定位的附属亚基,其表达异常会影响HCN通道的亚细胞分布和功能。因此,基于调节TRIP8b表达来影响HCN通道的表达水平及亚细胞定位,或基于TRIP8b在HCN通道上的作用位点,可以设计出影响HCN通道转运和定位的药物。此外,针对HCN通道磷酸化异常,可以设计能调节细胞内激酶活性或靶向HCN通道上激酶作用位点的药物,用以恢复HCN通道功能。现有的一些抗癫痫药物已表现出恢复HCN通道在海马中表达的作用,如抗癫痫药拉莫三嗪(lamotrigine)和加巴喷丁(gabapentin)通常被认为是钠通道阻断剂,但研究表明,它们同样可以恢复HCN通道在神经元树突上的表达,从而降低神经元兴奋性[44-45]。实验研究表明,对失神性癫痫鼠WAG/Rij进行早期干预,以恢复HCN通道的表达,同样也可降低皮层神经元兴奋性,减少癫痫的严重程度;而干扰神经限制性沉默因子(NRSF)与HCN1的相互作用,以增加HCN通道在潜伏期的表达,能明显推迟自发性癫痫的发作[46]。另有研究显示,靶向调节HCN通道的磷酸化水平,以改变其活性,对癫痫也有潜在治疗作用。在癫痫潜伏期,钙调磷酸酶抑制剂和p38激动剂均能增强HCN通道功能,对癫痫有不同程度的治疗作用[28]。
3 结语和展望
综上所述,HCN1和HCN2在脑内的表达和功能异常可直接影响神经元兴奋性,促使癫痫的发病。以HCN通道为靶点进行药物干预,已成为癫痫治疗领域的研究热点。因为人类心脏窦房结细胞主要表达HCN4亚基,所以非选择性HCN通道阻断剂,如ZD7288,会影响心脏正常起搏,具有心脏毒性。鉴于此,研发高选择性HCN1 或HCN2调节剂(增强剂或阻断剂)将成为癫痫治疗领域现阶段的研究重点。同时,由于缺乏特异性HCN通道调节剂,直接调节HCN通道的表达与功能,对动物的整体影响尚不十分明确。在实验研究中,HCN1基因敲除鼠未见明显发育缺陷,显示出认知能力增强的表型[47];HCN2基因敲除鼠有失神性癫痫表型[42],而利用病毒转染增加HCN2在下丘脑的表达,可明显减少癫痫发作[17]。但HCN1过表达鼠的表现尚未见报道。
目前,在HCN通道靶向治疗领域仍存在一些亟需探讨的问题:1)HCN通道在抑制性神经元中发挥什么功能,而阻断或增强抑制性神经元中HCN通道,对神经网络兴奋性会产生什么影响;2)在癫痫病人中已发现有HCN1基因突变,这些突变影响了HCN1功能,而HCN1基因敲除鼠虽然癫痫易感性增加,但并不出现自发性癫痫,其原因何在,是否意味着还有其他基因参与了癫痫的发生过程,这些基因为何以及与癫痫发生具有怎样的联系,又或者机体是否存在未被发现的神经保护机制,这些机制是否能够对癫痫的治疗产生作用;3)对以恢复HCN通道的功能与表达为靶向的药物研发,是否可以通过实时监测HCN通道的表达和功能而适时给药以及是否能够做到精准给药。这些问题的解答将会对HCN通道靶向药物研究与开发起到重要的推动作用。
[1]Löscher W, Klitgaard H, Twyman R E, et al.New avenues for antiepileptic drug discovery and development[J].Nat Rev Drug Discov, 2013, 12(10): 757-776.
[2]Staley K.Molecular mechanisms of epilepsy[J].Nat Neurosci, 2015, 18:367-372.
[3]Löscher W, Brandt C.Prevention or modifcation of epileptogenesis after brain insults: experimental approaches and translational research[J].Pharmacol Rev, 2010, 62(4): 668-700.
[4]Doeser A, Dickhof G, Reitze M, et al.Targeting pharmacoresistant epilepsy and epileptogenesis with a dual-purpose antiepileptic drug[J].Brain, 2015, 138(Pt2): 371-387.
[5]Robinson R B, Siegelbaum S A.Hyperpolarization-activated cation currents: from molecules to physiological function[J].Annu Rev Physiol, 2003, 65: 453-480.
[6]Santoro B, Chen S, Luthi A, et al.Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS[J].J Neurosci, 2000, 20(14): 5264-5275.
[7]Biel M, Wahl-Schott C, Michalakis S, et al.Hyperpolarization-activated cation channels: from genes to function[J].Physiol Rev, 2009, 89(3):847-885.
[8]Monteggia L M, Eisch A J, Tang M D, et al.Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain[J].Brain Res Mol Brain Res, 2000, 81(1/2): 129-139.
[9]Baruscotti M, Bottelli G, Milanesi R, et al.HCN-related channelopathies[J].Pfugers Arch, 2010, 460(2): 405-415.
[10]Lewis A S, Chetkovich D M.HCN channels in behavior and neurological disease: too hyper or not active enough? [J].Mol Cell Neurosci, 2011, 46(2): 357-367.
[11]Postea O, Biel M.Exploring HCN channels as novel drug targets[J].Nat Rev Drug Discov, 2011, 10(12): 903-914.
[12]Reid C A, Phillips A M, Petrou S.HCN channelopathies:pathophysiology in genetic epilepsy and therapeutic implications[J].Br J Pharmacol, 2012, 165(1): 49-56.
[13]Huang Z, Lujan R, Martinez-Hernandez J, et al.TRIP8b-independent traffcking and plasticity of adult cortical presynaptic HCN1 channels[J].J Neurosci, 2012, 32(42): 14835-14848.
[14]Huang Z, Walker M C, Shah M M.Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis[J].J Neurosci, 2009, 29(35): 10979-10988.
[15]Shah M M, Huang Z, Martinello K.HCN and K(V)7 (M-) channels as targets for epilepsy treatment[J].Neuropharmacology, 2013, 69: 75-81.
[16]Nava C, Dalle C, Rastetter A, et al.De novo mutations in HCN1 causeearly infantile epileptic encephalopathy[J].Nat Genet, 2014, 46(6): 640-645.
[17]Chung W K, Shin M, Jaramillo T C, et al.Absence epilepsy in apathetic, a spontaneous mutant mouse lacking the h channel subunit, HCN2[J].Neurobiol Dis, 2009, 33(3): 499-508.
[18]Pitkänen A, Sutula T P.Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy[J].Lancet Neurol, 2002, 1(3): 173-181.
[19]Huang Z, Lujan R, Kadurin I, et al.Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses[J].Nat Neurosci, 2011, 14(4): 478-486.
[20]Grone B P, Baraban S C.Animal models in epilepsy research: legacies and new directions[J].Nat Neurosci, 2015, 18(3): 339-343.
[21]Shah M M, Anderson A E, Leung V, et al.Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons[J].Neuron, 2004, 44(3): 495-508.
[22]Jung S, Jones T D, Lugo J N Jr, et al.Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy[J].J Neurosci, 2007, 27(47): 13012-13021.
[23]Marcelin B, Chauvière L, Becker A, et al.h channel-dependent defcit of theta oscillation resonance and phase shift in temporal lobe epilepsy[J].Neurobiol Dis, 2009, 33(3): 436-447.
[24]Chen K, Aradi I, Thon N, et al.Persistently modifed h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability[J].Nat Med, 2001, 7(3): 331-337.
[25]Powell K L, Ng C, O'Brien T J, et al.Decreases in HCN mRNA expression in the hippocampus after kindling and status epilepticus in adult rats[J].Epilepsia, 2008, 49(10):1686-1695.
[26]Brewster A, Bender R A, Chen Y, et al.Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specifc manner[J].J Neurosci, 2002, 22(11): 4591-4599.
[27]Shin M, Brager D, Jaramillo T C, et al.Mislocalization of h channel subunits underlies h channelopathy in temporal lobe epilepsy[J].Neurobiol Dis, 2008, 32(1): 26-36.
[28]Jung S, Bullis J B, Lau I H, et al.Downregulation of dendritic HCN channel gating in epilepsy is mediated by altered phosphorylation signaling[J].J Neurosci, 2010, 30(9): 6678-6688.
[29]Zha Q, Brewster A L, Richichi C, et al.Activity-dependent heteromerization of the hyperpolarization-activated, cyclic-nucleotide gated (HCN) channels: role of N-linked glycosylation[J].J Neurochem, 2008, 105(1): 68-77.
[30]Shah M M, Hammond R S, Hoffman D A.Dendritic ion channel traffcking and plasticity[J].Trends Neurosci, 2010, 33(7): 307-316.
[31]Lai H C, Jan L Y.The distribution and targeting of neuronal voltagegated ion channels[J].Nat Rev Neurosci, 2006, 7(7): 548-562.
[32]Yu H, Wu J, Potapova I, et al.MinK-related peptide 1: A beta subunit for the HCN ion channel subunit family enhances expression and speeds activation[J].Circ Res, 2001, 88(12): E84-87.
[33]Kimura K, Kitano J, Nakajima Y, et al.Hyperpolarization-activated, cyclic nucleotide-gated HCN2 cation channel forms a protein assembly with multiple neuronal scaffold proteins in distinct modes of proteinprotein interaction[J].Genes Cells, 2004, 9(7): 631-640.
[34]Qu J, Kryukova Y, Potapova I A, et al.MiRP1 modulates HCN2 channel expression and gating in cardiac myocytes[J].J Biol Chem, 2004, 279(42): 43497-43502.
[35]Gravante B, Barbuti A, Milanesi R, et al.Interaction of the pacemaker channel HCN1 with flamin A[J].J Biol Chem, 2004, 279(42): 43847-43853.
[36]Vasilyev D V, Barish M E.Regulation of the hyperpolarization-activated cationic current Ih in mouse hippocampal pyramidal neurones by vitronectin, a component of extracellular matrix[J].J Physiol, 2004, 560(Pt3): 659-675
[37]Lewis A S, Vaidya S P, Blaiss C A, et al.Deletion of the hyperpolarization-activated cyclic nucleotide-gated channel auxiliary subunit TRIP8b impairs hippocampal Ih localization and function and promotes antidepressant behavior in mice[J].J Neurosci, 2011, 31(20):7424-7440.
[38]Kurz J E, Sheets D, Parsons J T, et al.A significant increase in both basal and maximal calcineurin activity in the rat pilocarpine model of status epilepticus[J].J Neurochem, 2001, 78(2): 304-315.
[39]Sanchez R M, Dai W, Levada R E, et al.AMPA/kainate receptormediated downregulation of GABAergic synaptic transmission by calcineurin after seizures in the developing rat brain[J].J Neurosci, 2005, 25(13): 3442-3451.
[40]Much B, Wahl-Schott C, Zong X, et al.Role of subunit heteromerization and N-linked glycosylation in the formation of functional hyperpolarization-activated cyclic nucleotide-gated channels[J].J BiolChem, 2003, 278(44): 43781-43786.
[41]Brewster A L, Bernard J A, Gall C M, et al.Formation of heteromeric hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in the hippocampus is regulated by developmental seizures[J].Neurobiol Dis, 2005, 19(1/2): 200-207.
[42]Ludwig A, Budde T, Stieber J, et al.Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2[J].EMBO J, 2003, 22(2): 216-224.
[43]Budde T, Caputi L, Kanyshkova T, et al.Impaired regulation of thalamic pacemaker channels through an imbalance of subunit expression in absence epilepsy[J].J Neurosci, 2005, 25(43): 9871-9882.
[44]Poolos N P, Migliore M, Johnston D.Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites[J].Nat Neurosci, 2002, 5(8): 767-774.
[45]Surges R, Freiman T M, Feuerstein T J.Gabapentin increases the hyperpolarization-activated cation current Ih in rat CA1 pyramidal cells[J].Epilepsia, 2003, 44(2): 150-156.
[46]McClelland S, Flynn C, Dubé C, et al.Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy[J].Ann Neurol, 2011, 70(3): 454-464.
[47]Nolan M F, Malleret G, Dudman J T, et al.A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons[J].Cell, 2004, 119(5): 719-732.

[专家介绍] 黄卓:北京大学特聘研究员,博士生导师。2002年毕业于北京大学药学院。2008年5月获英国伦敦大学学院(University College London, UCL)神经药理学博士学位。2008.5-2013.8在UCL药学院药理系从事博士后研究员工作; 2013年9月入选教育部新世纪人才支持计划并同时入选北京大学青年人才“百人计划”。主要从事难治性癫痫的发病机制研究。其研究主要关注在神经元微小结构(树突、轴突和神经末梢)中离子通道在癫痫发病过程中表达、分布及其调控的变化。先后在Nature Neuroscience、Neuron、Journal of Neuroscience等期刊上发表学术论文12篇。
Progress in Research of Hyperpolarization-activated Cation Non-selective Channels and Antiepileptic Drugs Targeting the Channels
SUN Zhiming1,2, HUANG Zhuo1,2*(1.State Key Laboratory of Natural and Biomimetic Drugs, Department of Molecular and Cellular Pharmacology, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China;2.State key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China )
Epilepsy is a common neurological disorder characterized mainly by synchronous abnormal fring of a large number of neurons.It is now widely accepted that the imbalance between the excitatory and inhibitory synaptic transmission is the underlying pathogenesis of epilepsy.One third of epilepsy patients failed to response to the available treatments which mainly target sodium channels, calcium channels, potassium channels, glutamate receptors and GABA channels.Thus, the new targets and new ideas are urgently needed for antiepileptic drug development.More and more researches have suggested that the single-point mutation in hyperpolarization-activated cation non-selective channels(HCN channels) is able to induce genetic epilepsy.In addition, the changes in expression level, biophysical properties and subunit composition of HCN channels after brain injury contribute to epileptogenesis.Therefore, HCN channels and antiepileptic drugs targeting the channels have drawn much attention in recent years.In this article, the changes in HCN channels in epilepsy were summarized and the feasibility of antiepileptic drug development and treatment targeting HCN channels was discussed.
hyperpolarization-activated cation non-selective channel; epilepsy; antiepileptic drug
R966; R971.6
A
1001-5094(2015)02-0098-07
接受日期:2015-01-09
项目资助:国家自然科学基金面上项目(No.81371432)
*通讯作者:黄卓,教授,博士生导师;
研究方向:难治性癫痫发病机制研究;
Tel:010-82805925;E-mail:huangz@hsc.pku.edu.cn
