新一代测序的拷贝数变异检测算法研究与设计
2015-01-09李垚垚哈尔滨医科大学大庆校区黑龙江大庆163319
李 燕,李垚垚(哈尔滨医科大学大庆校区,黑龙江大庆163319)
doi:10.3969/j.issn.1672-5565.2015.03.07
新一代测序的拷贝数变异检测算法研究与设计
李 燕∗,李垚垚
(哈尔滨医科大学大庆校区,黑龙江大庆163319)
基于不同的测序技术,基因拷贝数变异的检测方法有多种,但时间复杂度较高,而新一代测序技术的发展为基因拷贝数变异检测的研究开辟了新领域。通过仿真实验、置换检验设计出一种新的基于新一代测序的拷贝数变异检测算法。不同于其它算法,本算法无需参考样本,通过直接研究比对后的序列以及reads与拷贝数的关系,来研究检测拷贝数变异,实验结果表明在时间复杂度上能提高50%以上的运算速度,这对今后拷贝数与疾病的研究具有重要意义。
新一代测序;拷贝数变异;仿真;置换检验
LIYan∗,LIYaoyao
(Harbin Medical University Daqing campus,Daqing Heilongjiang 163319,China)
新一代测序(New generation sequencing,NGS)技术的发展越来越成熟,各测序平台层出不穷,基因序列的测序成本大幅度地下降,测序的速度越来越高,这使得测序产生的DNA序列数据非常庞大,怎样理解数据成为当务之急。
伴随着人类基因组计划及 1 000 genomes project的实施与发展,蛋白质、DNA、RNA的序列数据的规模日趋增加,仅仅依靠生物实验来研究生物基因变异及疾病产生早已不能满足现实需要,因此必须借助计算机、数学等学科的理论及思想方法从海量数据中来研究和阐明生物学问题。拷贝数变异(Copy number alterations,CNAs)检测是生物信息学中研究生物基因结构改变的有效方法之一。
迄今为止,在HapMap计划的样本研究基础上,已经基本构建成人类第一代基因组CNV图谱[1]。随着测序技术的发展,新一代测序技术更成熟,从NGS数据出发,更多的拷贝数变异可能被检测,这也为研究CNV检测算法开辟了新领域。
新一代测序技术在对数据的处理过程中,会产生许多数据格式:FASTQ文件、SAM文件、VCF (Variant call format)文件、TXT文件和 BED文件等[2]。本文算法的重点研究对象是txt文件,当利用samtools工具中mpileup命令处理数据时,无“-g”或“-u”参数时会输出类似“.txt”文本文件,此文本文件统计了参考序列上每一碱基位点的比对结果,每一行表示reference中某一碱基位点的比对情况[3,14]。
1 拷贝数变异概述
1.1 拷贝数变异含义
诱发基因变异的因素有多个方面,基因的遗传变异的方式也多种多样[4]。大部分研究都表明,CNV指大小从Kb到Mb范围内的亚微观(Submicroscopic,指的是在普通电子显微镜下能分辨的范围)片段发生了拷贝数突变,这些拷贝数的复制、缺失、倒置等变异,统称为拷贝数变异(Copy number alterations,CNAs),但不包括转座子的插入和缺失引起的基因变异[5-7](见图1)。

图1 基因组中的拷贝数变异Fig.1 Copy number variation in genome
1.2 目前检测方法
目前拷贝数变异的检测方法主要分为三大类:一是定量PCR技术;二是 基于芯片的 array⁃based comparative genomic hybridization和SNPs芯片;三是新一代测序技术。
对于目标基因CNV检测常常采取基于定量PCR技术和杂交技术的方法。其中荧光定量PCR技术应用比较广泛,它的一个反应只测得一个拷贝,通过将检测样本的目标基因与参照基因定量后的检测值的比值相比较来估计此样本基因的拷贝数[6]。
基于芯片技术的CNV检测方法主要有:比较基因组杂交(Comparative genomic hybridization,CGH)技术、aCGH技术、oaCGH技术和SNPS芯片技术。其中,aCGH是基于微阵列的CGH技术,其芯片探针可以覆盖整个基因组,因此这种高通量分析法的准确度、敏感度和分辨度更高,结果更加准确[8]。SNPs芯片技术不同于CGH技术,仅仅使用单杂交就可实现检测。它是通过被测试的样本信号强度跟其他样本个体的强度作比较来确定每一位点对应的基因拷贝数[9]。这些方法都比较适合在全基因组范围内寻找CNV。
目前基于新一代测序数据的CNV方法主要有:分解读段(Split read)、读段深度(Read depth,RD)、末端配对法(Pair⁃end mapping,PEM)和重组(Assembly)等。由于新一代测序技术具有高通量、门槛低、简单等特点,因此基于NGS的CNV检测方法克服了杂交固有的某些缺点,即不需要太多特别复杂的设计工作,可以直接处理比对后数据,无需参考样本并可应用自身测序鉴定基因变化,而且费用相对低于aCGH技术。所以基于新一代测序的拷贝数检测方法具有良好的发展前景,这也为本次论文的研究内容提供了方向。
2 算法介绍
本文算法的目的是检测基于新一代测序的拷贝数变异,无需参考样本,这既减少了实验样本数量,还降低实验成本与时间。实验分为两大部分:(1)算法的设计及仿真实验;(2)真实数据的应用。
2.1 仿真实验
2.1.1 检验标准
在新一代测序时,高通量测序仪器一个反应得到的测序序列片段称为reads[10]。不同的测序仪器产生的reads数长度也不同,reads数的长度大小在36~200 bp不等。正常在没有发生拷贝数变异时,当测序depth和coverage一定时,同一测序仪器测序得到的一条染色体上的碱基序列上的reads数是基本相同的,若该序列上的reads数有一段区域不同于其他大部分区域,则可能说明这段reads数异常区域可能发生了拷贝数变化[11,15]。Reads数的异常主要表现在拷贝数的缺失、扩增等。因此本实验选取reads数作为衡量是否发生拷贝数的标准[15]。为了产生模拟数据这里自行定义reads数S=40bp,为测得正常序列的reads。若测序区域<40或>40,我们都认为其发生了拷贝数变异。
2.1.2 仿真数据
由于受到目前测序仪器和水平的限制,测序所得碱基序列的reads数会不一致,reads数可能会上下波动,但仍然处于相同水平。这里选reads∈[39,40,41]来模拟实验数据。
Simulation的过程:
(1)随机构建一个染色体位点数为2 000的样本,并对每个位点编号。
(2)任取多个区域如100-149,500-529,900-919,1 600-1 650,对其进行信号加强/减弱处理,模拟成这几段标记区域发生reads数变化(即拷贝数改变)[12-13]。
为了使实验数据更逼近实际测得序列,减少误差,需要对仿真数据进行加噪声处理。这里主要是利用高斯噪声处理,并对随机其他位点噪声处理。
经过上述步骤,产生了一个包含2 000个位点的样本。而在统计实验中一个样本不能证明任何实验问题,需要大量的样本才能减少误差,得出结论。因此我们重复上述步骤,产生了50个样本用于实验。
2.2 置换检验
2.2.1 置换检验概述
通常显著性检验可以确定一个观测值是否有效[16],如假设检验中检测两组样本的均值是否有相等(或者检测哪一均值更大)。本次实验仿真出一些小样本结果(这里是50个小样本),借助于Permutation test置换检验来分析小样本的总体分布。
Permutation test是20世纪30年Fisher提出的基于大量计算,根据对样本中的数据随机(或全)排列,统计并推断的一种方法。算法公布之初,由于它的运算量没能得到重视与应用。近年来随着计算机的性能提高,我们可以借助计算机的计算能力来实现置换检验来解决问题。它是基于样本本身的,对样本的总体分布要求自由,因此应用相对较广泛,尤其适合用于对总体分布未知的小样本数据分析,以及一些用常规方法难以分析的假设检验问题。置换检验的过程一般是:首先对样本内的数据进行顺序置换,然后重新计算检验统计量,并构造出经验分布,最后求出P⁃value来推断结果。
2.2.2 算法设计与实现
假设设计一个实验来验证仿真实验中样本位点数100-149,500-529,900-919,1 600-1 650的区域发生了拷贝数改变。(实验数据被保存在merge1.txt中。)
用假设检验的方法完成验证,选取样本位点对应的reads数构造为检验统计量。零假设为:样本碱基对应的reads数没有发生改变(即碱基序列对应的copy number未发生改变,是正常的)。在这个检验中,最终计算出2 000个位点对应的p⁃value值若<0.05的区域,则表明小概率事件发生,而原假设是正常的,因此原假设错误,此区域(位点)发生了拷贝数变异。
采用置换实验,计算每个位点对应的p⁃value(P [j],j=1,2,…,2 000):
P[j]=f[j]/1 000
其中,f[j]为每个位点对应的频数。
观察每个位点对应的p⁃value,并绘制见图2。
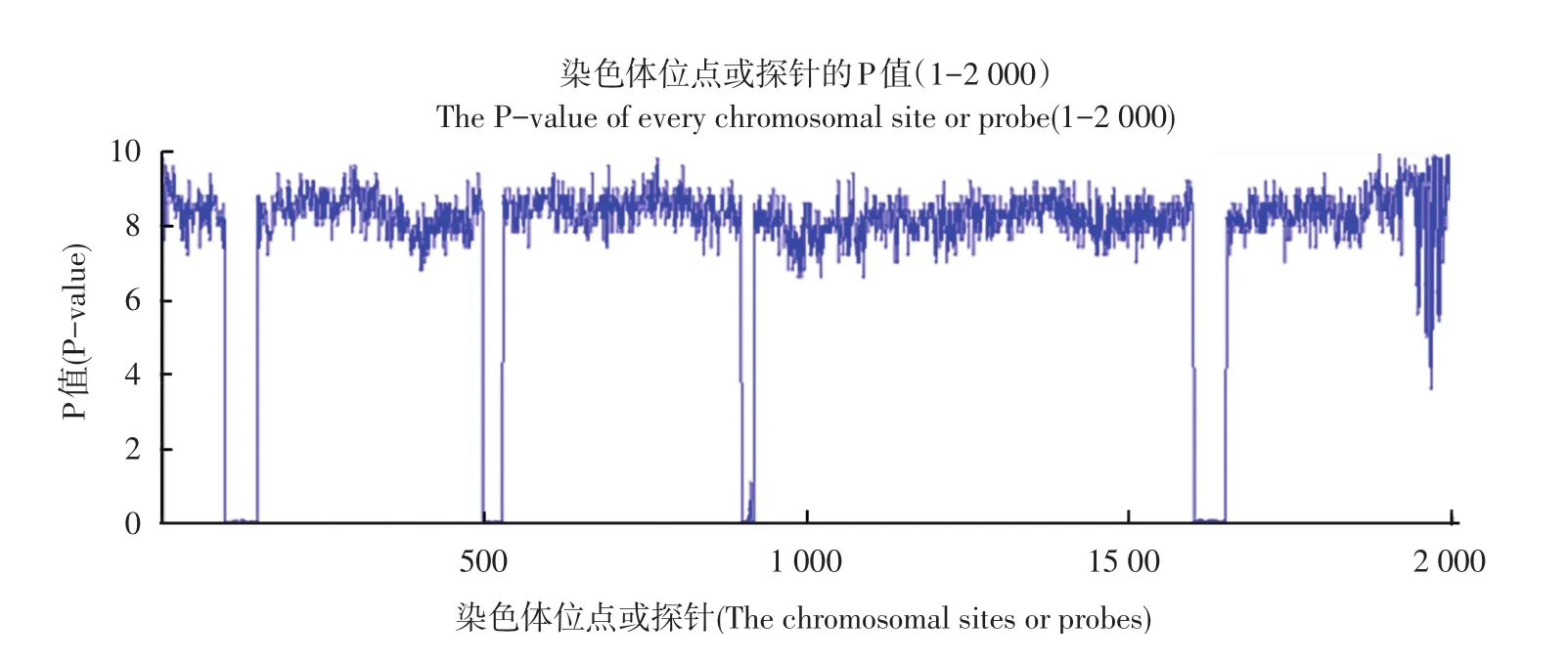
图2 各位点对应P值Fig 2 The p⁃value of every site
2.2.3 实验结论
由图1放大可以直观看出在100-149,500-529,900-919,1 600-1 650区域的p⁃value值大小明显<0.01,说明在这些区域小概率事件发生,原假设错误,而是在这些区域发生了拷贝数变异。这与仿真数据时的变异区域相同,因此本算法可以检测拷贝数变异。
3 真实数据CNV检测
3.1 数据来源与处理
为检测上述方法的适用性,本文从 1 000 genomes project数据库中获得真实数据,为了保证数据可用性,下载真实数据要确保控制单一变量reads数变化,其他如read depth、read coverage、测序仪器等要控制一致[17]。这里采用 HG00096. mapped.ILLUMINA.bwa.GBR.low_coverage.20120522. bam.中chrom20的数据作为数据应用上述检测方法。同时为了证明在high coverage数据同样适用,还处理了 HG00096.mapped.ILLUMINA.bwa.high_coverage.bam数据。
利用Samtools软件对真实数据进行处理,CBS方法去除噪声,提取reads数,统计频数最多的reads值。考虑真实数据噪声和测量误差,可确定实验数据区域正常情况下reads数在[39,43],并以此为基准检测该区域内是否发生了拷贝数变异。若区域内位点对应的reads值小于或大于这个区间,认为对应位点发生了拷贝数变异。
3.2 CNV值计算
正常情况下,人类基因拷贝数变异的值为2。研究表明,某一位点拷贝数变异的数目与对应的reads值成如下的关系[19]:其中R0为测序深度、覆盖度一致时正常情况下区域或位点对应的reads值,R1为待测区域或位点对应的reads值,x即为待测区域或位点的拷贝数的值。因此可以计算任意位点的拷贝数。
2/R0=x/R1
3.3 数据结果分析
本实验HG00096.mapped.ILLUMINA.bwa.GBR. low_coverage.20120522.bam.中chrom20上的seq1:1-1 569位点和seq2:37-1 567位点上的reads数据应用上述算法,并绘制如下图3。图3为chrom20的seq1:1-1 569和seq2:37-1 567位点对应的reads数分布情况,图4和图5分别为seq1和seq2相应位点reads数分布图,其中红线部分表示被测区域内reads数出现最多的数值。大多数位点都在红线附近上下波动,当位点对应的reads数距离红线越远时,我们认为该位点可能发生了拷贝数变异。如图5中seq1:1-220点附近,图5中seq2:190-250位点附近等,我们可以很直观地推测这些区域可能发生了拷贝数变异。还可根据数据确定变异边界,利用公式计算各位点对应的拷贝数值。
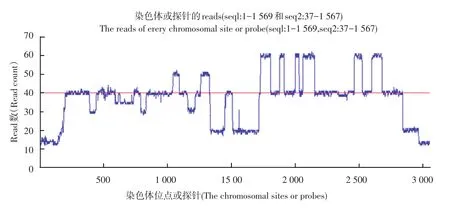
图3 Chrom20 seq1:1-1 569和seq2:37-1 567上位点对应的reads数分布图Fig.3 Reads distribution map of the sitechrom20 seq1:1-1 569 and seq2:37-1 567

图4 Chrom20 seq1:1-1 569上位点对应的reads数分布图Fig.4 Reads distribution map of the sitechrom20 seq1:1-1 569

图5 Chrom20 seq2:37-1 567上位点对应的reads数分布图Fig.5 Reads distribution map of the site chrom20 seq2:37-1 567
3.5 算法的性能与评价
3.5.1 仿真代码实现上
本算法程序代码基于R语言相对容易实现,对于涉及的数据预先分配空间,大大降低了时空复杂度。但是在permutation、merge data以及做test时会涉及到双层for循环,再加之数据样本自身很大,因此增加了时间复杂度。为了减少时间消耗,提升速度,在编写代码时除采用了向量化避免for循环,加入并行运算方法。
3.5.2 算法应用上
算法基于新一代测序技术测序数据,与基于芯片的检测技术相比,本算法无需参考样本,数据来源更真实,使得检测的拷贝数也更真实,大大减少了误差,同时也最大的降低了检测费用。
本算法在双核x86 32 bit的处理器中执行,经检验,在内存占有量相差无几时,时间复杂度降低明显(本算法样本计算时间120.2 s,CNV-seq计算时间251.5 s,,FREEC计算时间319.6 s),如图6所示。同时在检测边界也具有相当高的灵敏度,直接从比对后的数据处理,也降低了从raw data到mapped data中产生的各种误差。
本算法能够检测出拷贝数变异,但是对拷贝数变异的类型不能很清晰的界定,这一方面有待改善。它对测序数据的格式等要求比较严格,要保证实验数据序列的read coverage,read depth等一致,还要保证数据是基于同一测序技术测得的。同时,它只对新一代测序的数据有效,随着第三代测序技术的萌芽,在检测拷贝数变异时可能会出现瑕疵,但可以借鉴思想,在未来很长时间仍然受用无穷。
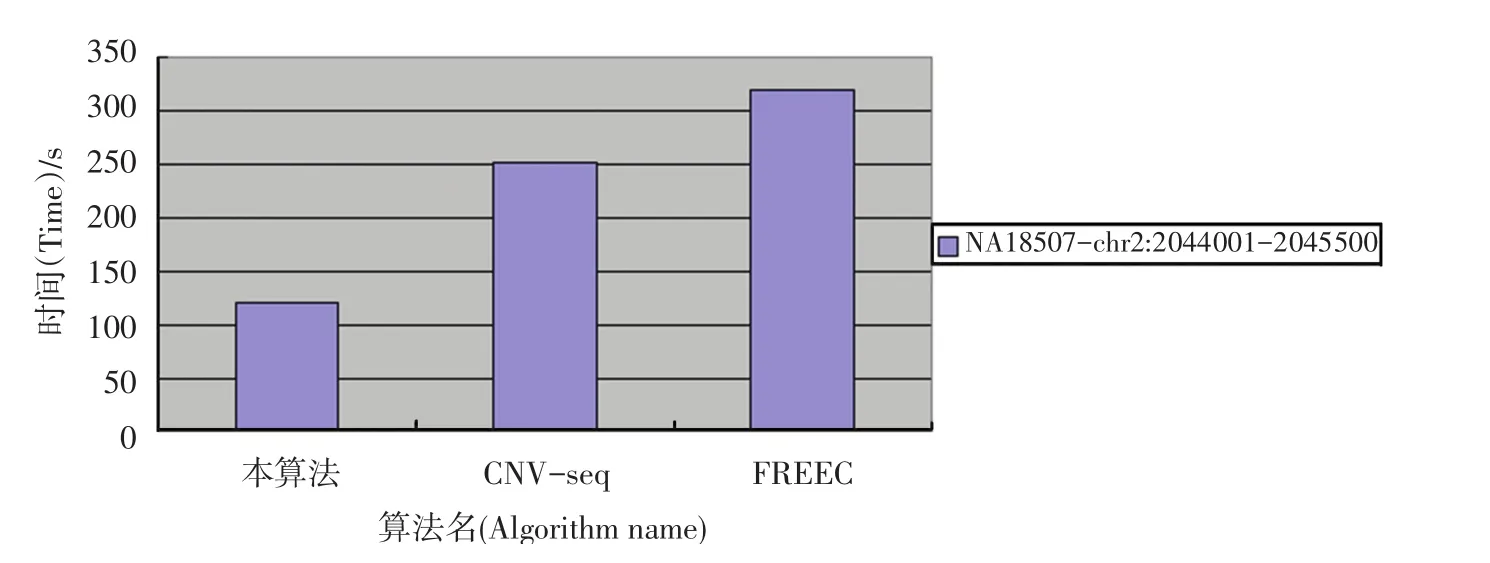
图6 算法时间复杂度对比Fig.6 Algorithm time complexity contrast
4 结束语
CNV作为基因结构变异的一种重要形式,对人类遗传进化、疾病和药物研究等具有重要的意义[20]。在众多检测方法中,目前急需效率高和准确性高算法。本文提出了一种基于新一代测序数据的CNAs检测算法,无需额外的参考样本序列,利用置换检验的方法检验,降低假阳性率,增强结论的真实性,提高了准确度。实验表明,这种基于新一代测序的拷贝数变异检测算法,可快捷方便地找出由新一代测序技术测得的染色体上可能发生拷贝数变异的位点,大大降低了时间复杂度。这对今后拷贝数与疾病的研究具有重要意义。
[1] FREEMAN J L,PERRY G H,FEUK L,et al.Copy number variation:new insights in genome dicersity[J]. Genome Res,2006,16:949-961.
[2] SHENDURE J,JIH.Next⁃generation DNA sequencing [J].Nat Biotechnical,2008,26:1135-45.
[3] SCHUSTER SC.Next⁃generation DNA sequencing transforms today's biology[J].Nat Methods,2008,5:16-8.
[4] IAFRATE A J,FEUK L,RIVERA M N,et al.Detec⁃tion of large⁃scale variation in the human genome[J].Nat Genet,2004,36(9):949-951.
[5] XIE C,TAMMIM T.CNV⁃seq,a new method to detect copy number variation using high⁃throughput sequencing [J].BMC Bioinformatics,2009,10:80.
[6] BOEVA V,ZINOVYEV A,BLEAKLEY K,et al.Con⁃trol⁃freecalling of copy number alterations in deep⁃sequen⁃cing data using GC⁃contentnormalization[J].Bioinformat⁃ics,2011,27(2):268-269.
[7] REDON R,ISHIKAWA S,FITCHK R,et al.Global variation in copy number in the human genome[J]. Nature,2006,444:444-454.
[8] COOPER G M,NICKERSON D A,EICHLER E E. Mutational and selective effects on copy⁃number variants in the human genome[J].Nature Genetics,2007,39:S22-29.
[9] CHIANG D Y,GETZ G,JAFFE D B,et al.High⁃resolution mapping of copy⁃number alterations with massively parallel sequencing[J].Nat Methods,2008,6 (1):99-103.
[10]MILLER C A,HAMPTON O,COARFA C,et al. ReadDepth:a parallel R package for detecting copy number alterations from short sequencing reads[J].PLoS ONE,2011,6:16327.
[11]YOON S,XUAN Z,MAKAROV V,et al.Sensitive and accurate detection of copy number variants using read depth of coverage[J].Genome Res,2009,19:1586-1592.
[12]VANCE A.Data analysts captivates by R's power[J]. New York Times,2009,6:22-29.
[13]VENABLESW N,SMITH D M,TEAM R D C.An introduction to R[M].Network Theory,2006:34-38..
[14]LIH,HANDSAKER B,WYSOKERA,et al:The sequence alignment/map format and SAMtools[J]. Bioinformatics,2009,25(16):2078-2079.
[15]MEDVEDEV P,FIUME M,DZAMBA M,et al. Detecting copy number variation with mated short reads [J].Genome Res,2010,20(11):1613-1622.
[16]MAGIS,TATTINI L,PIPPUCCI T,et al.Read count approach for dna copy number variants detection[J]. Bioinformatics,2012,28(4):470-478.
[17]WANG J,WANG W,LI R,et al.The diploid genome sequence of an Asian indicidual[J].Nature,2008,456:60-65.
[18]TIERNEY L,ROSSINI A J,LI N.Snow:A parallel computing framework for the R system[J].Int JParallel Program,2009,37(1):78-90.
[19]ABYZOV A,URBAN A E,SNYDER M,et al.An approach to discover,genotype,and characterize typical and atypical cnvs from family and population genome sequencing[J].Genome Res,2011,21(6):974-984.
[20]KORBEL JO,URBAN A E,AFFOURTIT J P,et al. Paired⁃end mapping reveals extensive structural variants detection in the human genome[J].Science,2007,318 (5849):420-426.
An algorithm for detecting copy number alteration from next generation sequencing of human genome
Based on different sequencing technologies,the detection methods of gene copy number variation are available.However,with the development of new generation sequencing technology,a new field for researchingcopy number variations has been opened up.Through the simulation experimentand the replacement test,this paper designs a new copy number variation detection algorithm based on the new generation of sequencing.Unlike other algorithms,our algorithm doesn't need thereference samples,butuses themapped datafrom next generation sequencing platforms and the relationship between reads and gene copy number to detect gene copy number variations in the genome.The experimental results show that the performance in time complexity can be improved bymore than 50%,indicating the important significance for the further study of gene copy number and disease in the future.
Next generation sequencing;Copy number variations;Simulation;Permutation test
TP301.6
A
1672-5565(2015)03-186-06
2015-06-19;
2015-07-14.
黑龙江省教育厅科学技术研究项目(12541565)。
李燕,女,教授,研究方向:数据库与数据挖掘;E⁃mail:qliyan@163.com.
